Medical device nomenclatures are those products used to prevent, diagnose, treat, and monitor the many diseases known to humankind. Medical devices and medicines play an equally important role in treating human beings.
To learn more about medical devices, read our article on the definition of a medical device. This article discusses the nomenclature of medical devices and examples of these.
What is the Nomenclature of Medical Devices?
To simply put it, the nomenclature is the naming of a medical device. Although medical devices are classified into different risk classes, they should be named so that it is universally identified. Standardised nomenclature facilitates this easy identification.
A medical device nomenclatures is needed to simplify trade and tracking among the different regulatory authorities, Ministries of Health, and other organisations that regulate medical devices.
A standardised medical device nomenclatures aids in the following aspects:
Grouping and classification of medical devices.
Registration under different regulatory bodies or Ministries of Health
Streamlined procurement and distribution
Grouping of medical devices in various electronic health records and medical device databases
Vigilance reporting, field safety and post-market surveillance
The different medical device nomenclatures available are as follows:
GMDN or Global Medical Device Nomenclatures.
EMDN or European Medical Device Nomenclatures
UMDNS or Universal Medical Device Nomenclatures System
Other nationally developed nomenclature systems
Global Medical Device Nomenclatures (GMDN)
About 10% of countries use Global Medical Device Nomenclatures worldwide. It is a system of internationally accepted descriptors used to identify medical devices. The GMDN Agency manages GMDN codes, a non-profit organisation.
GMDN is a 5-digit code containing the following information:
GMDN Term Name: Anaesthesia ventilator
GMDN Code: 34851
GMDN Definition: A mains electricity (AC-powered) stand-alone, automatic cycling device used to assist and control alveolar ventilation during general anaesthesia and is compatible with inhaled anaesthetic agents. It has fewer functions and is less complex to operate than an intensive care ventilator but adequately meets the patient’s ventilation needs for oxygen (O2) and carbon dioxide (CO2) exchange to maintain normal blood gas concentrations. The device provides a mechanical means to deliver the breathing gas to the patient in a controlled pattern. It is equipped with alarms to warn of changes in respiration or the onset of unsafe operating conditions.
GMDN was introduced for a variety of regulatory purposes. GMDN is based on the ISO 15225: Medical device nomenclature data structure’. Read more about the frequently asked questions about GMDN here.
European Medical Device Nomenclature (EMDN)
European Medical Device Nomenclature or EMDN is introduced due to Article 26 of EU Regulation 2017/745 of Medical devices and Article 23 of EU Regulation 2017/746 of in-vitro diagnostic medical devices.
Like GMDN, it plays a considerable role in device nomenclature and serves various regulatory purposes. One of the primary uses is while registering a medical device in EUDAMEDwhere it is closely linked to UDI-DI.
Structure of EMDN
The European Medical Device Nomenclature is characterised by its alphanumeric structure and is established in a seven-level hierarchical tree where it clusters medical devices into three primary levels:
Categories: the first hierarchical level – alphanumeric.
Groups: the second hierarchical level – 2 numbers indicating group.
Types: the third hierarchical level – a series of numbers 1,2,3,4 and 5.
EMDN was adopted from the Classificazione Nazionale Dispositivi medici (CND) classification. The EMDN can be accessed at the EMDN list. European Medical Device Nomenclature categorises into three primary levels, categories, groups, and types.
A category comprises several groups composed of various kinds of medical devices.
Source: https://webgate.ec.europa.eu/dyna2/emdn/. In the above image of EMDN, ‘A’ is the category, ‘A01’ is Group and ‘A0101’ to ‘A0199’ are the types of medical devices.
Universal Medical Device Nomenclature System (UMDNS)
Universal Medical Device Nomenclature System or UMDNS was developed by the Emergency Care Research Institute (ECRI). Many nations have adopted this standard around the world. UMDNS is used in inventory control, work order control, and regulatory systems applications.
It is a 5-digit code unique code and a term for different types of medical devices. One can find the UMDNS code list here. UMDNS is updated monthly.
CND Nomenclature
CND nomenclature or ‘Classificazione Nazionale Dispositivi medici’ was developed by Italian Ministry of Health. In addition to Italy, it is also used in Portugal and Greece.
The guidance document on CND nomenclature explains the basic principles and structure of CND, which also applies to EMDN as EMDN was adopted from CND nomenclature. Following this, medical devices are clustered into three levels:
Category
Group
Type
FAQs
Is UDI the same as GMDN?
Both Unique Device Identification System (UDI) and GMDN are used in device identification. However, the two have some fundamental differences. UDI is inferior because of its lack of unity. It does not have a structure; therefore, device identification becomes more difficult with UDIs. Nonetheless, it is an effective tool for the traceability of medical devices. FDA utilises UDIs for medical device identification. The user guide to GUDID explains how the UDIs are managed in US FDA’s database, GUDID.
Can EMDN be accessed free of charge?
The EMDN is accessible to all stakeholders- free of charge. Hence, it can be utilised by a non-exhaustive list of stakeholders such as manufacturers, patients, research organisations, practitioners, hospitals, etc. The EMDN can also be downloaded from here.
Is there a guidance document that helps economic operators to map the EMDN information into the forthcoming EUDAMED database?
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.
Good manufacturing practices are the procedures that must be followed in order to comply with the guidelines set forth by regulatory agencies that oversee the authorization and licencing of the manufacture and sale of medical devices.
In this article, we will discuss the Significant changes that were made in the latest 2022 Regulations, which supersedes the 2013 version.
In Art 35 2022 GMP Document, under the document review and archive section, it is explicitly mentioned that – “All digitally stored documents and records shall be backed up.”
In Art 71 2022 GMP Document, under the Personal health and hygiene section, it is now clearly explained that – “Any individual who, upon medical examination or observation by supervisors, appears to be in a health condition which could affect the product shall be removed from operations until the health condition is deemed adequate.”
In Art 87 2022 GMP Document underlabels and instructions for use, there is an additional point which states – “In the case of importers, the approval documentation referred to in “Labels and instructions for use” article may be recorded in a specific document in lieu of the history record of the product.”
In Art 112 2022 GMP Document under Distribution Records section, it is now mentioned as:
Each manufacturer shall maintain distribution records which include or reference:
I – the name and address of the consignee.
II – risk: combination of probability of occurrence and severity of damage.
III – any number control used for traceability.
Previously in the 2013 GMP document, it was mentioned as:
Each manufacturer shall maintain distribution records, including or making reference to: (i)Names and addresses of the consignee; (ii) Identification and amount of products shipped, with shipment date; (iii) Any numerical control used for traceability.
In Art 114 2022 GMP Document under Identification and traceability of batch production units’ section, there is an additional point added which states- “Each manufacturer shall establish and maintain a program for maintenance, adjustment and, where necessary, cleaning of equipment to ensure that all manufacturing specifications are met.” This is in addition to the identification of the units that are manufactured with every product history record.
In Art 118 2022 GMP Document, under the Non-conforming components and products section, there is an additional point added which states – “The disposal of the products referred to as “non-conforming” shall be documented, and a record of the justification and manual or electronic signature of the person-in-charge for the disposal of such non-conforming products or components shall be kept.”
In Art 120-VIII 2022 GMP Document, under the Corrective and preventive actions section, there is additional information added which states – Each manufacturer shall establish and maintain procedures to determine the collection of products and other field actions for those products that are already distributed. The quality process to determine the corrective actions in regard to the identified potential causes shall be based on a valid statistical technique for detecting recurring quality problems, where applicable.”
In Art 123 2022 GMP Document under the Quality Audit section, there is an additional point added which states – “Those in charge of conducting the quality audit cannot be directly responsible for the matters under audit.”
CHAPTER 10 in 2022 GMP Document (Final Provisions) is additionally introduced – The chapter includes rules such as:
“The documentation evidencing compliance with the requirements set out in this Resolution shall be made available whenever requested by the health regulatory agencies.”
“Failure to comply with the provisions contained in this Resolution constitutes a health violation, under the terms of Law No. 6.437, of August 20, 1977, without prejudice to applicable civil, administrative and criminal liabilities”
“The following are hereby revoked: I – the Resolution of the Collegiate Board of Directors – RDC No. 16, of March 28, 2013, published in the Federal Official Journal No. 61, of April 1, 2013, Section 1, p. 75;
II – Normative Instruction – IN No. 8, of December 26, 2013, published in the Federal Official Journal No. 252, of December 30, 2013, Section 1, p. 758.”
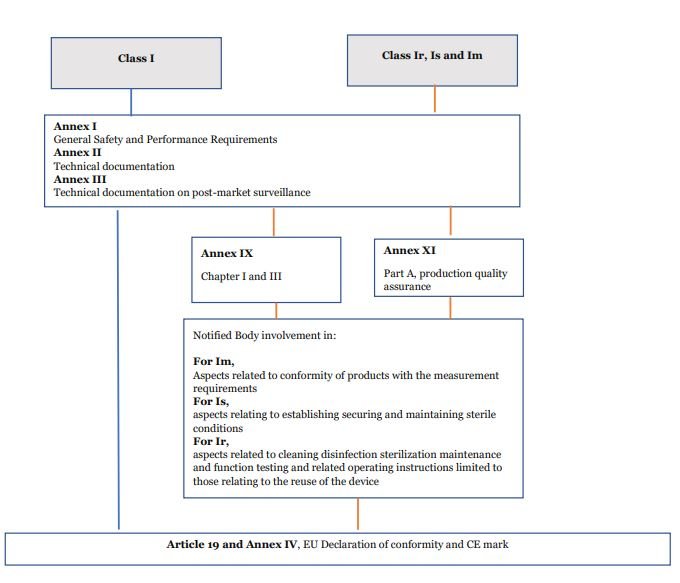
The EU MDR 2017/745 imposes more stringent requirements for Class I devices. Under the new regulations, Manufacturers must righty classify a medical device and provide technical documentation following the device class.
The risk class under MDD could change under MDR. In some cases, medical devices could be up classified from Class I medical devices to Class II a/b medical devices or Class III medical devices.
MDCG guidance on Class I medical devices is a valuable resource document for Class I medical devices Manufacturers. Manufacturers can place the devices by following the steps mentioned below.
Steps for placing a Class I Medical Devices
Step 1: Integrate MDR into the existing Quality Management System.
This allows the correct documentation to be created following the risk classification of the device.
Step 2: Confirm whether the product is a medical device under the scope of MDR.
Confirm that the product meets the definition of a medical device as stated in Article 2 of EU MDR based on its intended use and primary mode of action. In some cases, the product may be out of the scope of MDR and may be termed as a medical device ‘accessory ’. EU MDR also states how accessories for a medical device is also to be classified on its own apart from the main medical device.
Step 3: Confirm whether the medical device is a Class I medical devices.
It must be noted that several MDD Class I medical devices will be reclassified under the MDR as per the new classification criteria in Annex VIII, such as most software (rule11) and Class I medical devices made up of chemicals or combinations of substances (rule 21).
Step 4: Pre-market procedures.
Verify that the device meets the General Safety and Performance Requirements (GSPR).
A proper and well-established risk management system should be implemented. Manufacturers must ensure that the associated risks are identified, analysed and appropriate corrective actions are taken.
This must be guaranteed throughout the product’s lifecycle and documented. When standard specifications are available, Manufacturers must adhere to them unless they can demonstrate that they have adopted a solution that is at least as safe and effective.
The Manufacturer should periodically update the clinical evaluation report, risk management and post-market reports.
Conduct Clinical evaluations
All devices require clinical evaluation, Class I medical devices are not exempted from this requirement. A clinical evaluation report must contain essential information such as device descriptions, literature reports and post-market surveillance reports to name a few.
MDR also emphasises the need to consider available alternatives and the acceptability of the benefit-risk ratio.
Prepare an up-to-date technical file.
The Manufacturer will draw up the technical documentation that demonstrates the conformity of their devices with the specific requirements of the MDR. Technical documentation is mentioned in Annex II and III of EU MDR.
The Manufacturer is obligated to keep the file updated and make them available to competent authorities, notified bodies and the authorised representative.
The technical file must contain the following elements. The detailed information can be found in Annex II of EU MDR.
Device description and specification, variants, and accessories
Information to be supplied by the Manufacturer
Information on Design and manufacturing
General Safety and Performance Requirements
Benefit-risk analysis
Risk management
Verification and validation of product
Demonstration of Conformity
The documents in the technical file should be made available in the language accepted in the Member State in which the device is sold. Please read our article on EU Requirements for translations to understand this requirement specified in EU MDR.
Involvement of Notified Bodies
Most Class I devices do not require the involvement of a Notified Body. However, device Classes ‘Ir’, ‘Is’ and ‘Im’ have this requirement. ‘Ir’ devices are those which are reusable instruments. ‘Is’ are devices to be used in a sterile condition. ‘Im’ are those devices with a measuring function.
For these class I medical devices, the involvement of Notified Bodies is limited to audit and the NB assesses those specific controls. For example, an “Is” device assessment of NB would involve auditing the Sterilisation process and controls.
Prepare Instructions for Use and Labelling
Medical device manufacturers must ensure that the instructions for use and labels are available in languages accepted within the Member State they decide to market in.
The instruction for use must indicate the safety and performance information so that its users are aware of its intended use. The instructions for use (IFUs) should have drawings and internationally accepted symbols.
The Declaration of conformity (DoC) is a document signed by the Manufacturer that states the device fulfils the requirements set forth by MDR. For more information, read our article on Declaration of conformity.
Step 6: Affix CE mark.
CE mark should be placed on the medical device to show compliance. If it cannot be placed on the product itself, it must be placed on the packaging.
Before placing in the EU market, all devices must be registered on EUDAMED. EUDAMED is a secure, web-based European Databank on Medical Devices. Although Class I devices are not much of a threat to human life, they must be made available on EUDAMED. It ensures that the device information is accessible to the public.
Transitional provisions for Class I Medical Devices
Transitional provisions are in place to help manufacturers transition to MDR from the directives. Article 120 of EU MDR gives an idea of the exemptions from MDR and greatly helps manufacturers by providing more time to meet the requirements.
The following conditions must be met to be able to market a device according to Directive 93/42/EEC until 26 May 2024:
The device complies with Directive 93/42/EEC.
The device has not undergone significant changes in device design or intended purpose.
The device has a valid Declaration of Conformity (DoC)
The post-market surveillance, Market Surveillance, Vigilance and Registration of economic operators and devices requirements are met.
FAQs
Does a Class I Device manufacturer require an ISO certificate for the Quality Management System?
Not necessarily. The Class I manufacturer must have an established QMS, and obtaining a certification is not mandated. Although globally, there is a significant benefit for the Manufacturer to hold an ISO QMS certification. Major markets worldwide accept ISO 13485 certificate as one of the key entry factors into their geography. A few key markets, such as Canada, Japan, Australia, Singapore, and Malaysia, require ISO Certification. The ISO 13485 certificate is recognised worldwide as a significant standard for a quality management system for medical device manufacturers.
Should Class I Device DoC contain the reference to MDR Annex (s)?
Yes, but this has been considered of less importance while drafting the DoC. Declaration of Conformity (DoC) must mention the respective Annex(s) in the MDR that it has complied with for any device class. This is one of the required information to be entered while performing product registrations in many countries.
What is the conformity procedure for Class I devices?
The conformity procedure for all Class I devices is mentioned in the flow chart.
Annex XVI products are those for which a manufacturer claims only an aesthetic or another non-medical purpose but are like medical devices in terms of functioning and risk profile.
These categories of products were added in the new Regulation to establish production and surveillance standards for these previously unregulated products to protect users’ health and safety.
LIST OF GROUPS OF PRODUCTS WITHOUT AN INTENDED MEDICAL PURPOSE REFERRED TO IN ARTICLE 1(2)
Contact lenses or other items intended to be introduced into or onto the eye.
Products intended to be totally or partially introduced into the human body through surgically invasive means for the purpose of modifying the anatomy or fixation of body parts except for tattooing products and piercings.
Substances, combinations of substances, or items intended to be used for facial or other dermal or mucous membrane filling by subcutaneous, submucous or intradermal injection or other introduction, excluding those for tattooing.
Equipment intended to be used to reduce, remove, or destroy adipose tissue, such as equipment for liposuction, lipolysis or lipoplasty.
High-intensity electromagnetic radiation (e.g., infra-red, visible light and ultra-violet) emitting equipment intended for use on the human body, including coherent and non-coherent sources, monochromatic and broad-spectrum, such as lasers and intense pulsed light equipment, for skin resurfacing, tattoo or hair removal or other skin treatment.
Equipment intended for brain stimulation that apply electrical currents or magnetic or electromagnetic fields that penetrate the cranium to modify neuronal activity in the brain.
When is this coming into force?
Manufacturers of Annex XVI products have six months from the official release of the standard specifications, which is scheduled in Spring 2022, to comply with the regulation.
As a result, manufacturers must examine how this may affect their product(s) right now and what further testing and proof will be required to demonstrate compliance with common specifications.
Technical and clinical requirements (other than standards) that give a way of complying with the legal duties applicable to a device are referred to as “common specifications.” Existing harmonised standards for similar medical equipment will be considered.
Annex XVI products do not have to comply with the MDR until the Commission adopts and publishes the CS for the individual product categories, which is expected to be no later than May 26, 2020, or when CS is adopted, whichever is the latest.
Individual Member States’ national classification of Annex XVI products as medical devices will continue to apply until then, and compliance with applicable national medical device legislation will be required.
How will this impact manufacturers of these products?
Manufacturers of Annex XVI products sold in Europe (including the UK) will have to be compliant with the requirements laid out in the MDR for general medical devices to ensure they are safe for use.
In addition to complying with the relevant standard specifications, manufacturers of Annex XVI products will need to meet several other obligations such as GSPR, equivalence approach, clinical data, and post-market surveillance.
These include, but are not limited to, ensuring that:
Correct classification of the device based on the risk impact (as per Annex VIII)
Comply to the Common Specifications listed for those products
Require demonstrating clinical benefit in accordance with Annex XIV & annex XV
There is a person in charge of regulatory compliance
Distributors and importers in the supply chain are compliant
Sufficient financial coverage is in place in respect of a manufacturer’s potential liability
The new vigilance reporting deadlines have been met, and an annual safety update report has been issued.
Manufacturers must undergo a conformity assessment, which will be conducted by a notified authority, as part of the regulatory requirements.
The device must then be marked with a CE mark and a declaration of conformity. Furthermore, a basic unique device identification (UDI) must be assigned and provided to the UDI database, critical information about themselves, and the authorised representative and importer if they are based outside of the EU, needs to be submitted to EUDAMED.
The manufacturer must also comply with post-market surveillance and vigilance requirements, including reporting remarkable events to the appropriate authorities, such as the MHRA in the UK.
Manufacturers of Annex XVI products will have additional requirements, such as verifying that the device has been accurately classified against the revised risk classification criteria in Annex VIII of the MDR and complying with the necessary standard specifications.
They will also need to ensure they have a person in charge of regulatory compliance, have enough insurance to cover potential manufacturer liability, meet the new vigilance reporting deadlines, and generate an annual periodic safety update report.
The manufacturer is also responsible for ensuring that the product’s distributors and importers have performed their duties under Articles 13 and 14 of the regulation.
Please read Article 10 of the MDR for further detail on manufacturer obligations.
How to comply with the legal requirements?
Determine which risk category your device belongs to.
Ensure your device conforms with the MDR and the standard specifications for these groups of products.
Pass a compliance assessment conducted by a notified body if required.
Draw a declaration of conformity and affix a CE mark to the device.
Provide the UDI database with a basic unique device identification (UDI) assigned to the device.
If the manufacturer is outside the EU, submit essential information about the manufacturer, authorised representative, and importer to the electronic system (Eudamed).
Place your CE marked device on the market or put it into service anywhere in Europe.
Meet the standards for post-market surveillance and vigilance, such as implementing field safety remedial actions and reporting major events to the appropriate authority.
Examples of Annex XVI products
Non-prescription coloured contact lenses
Solid contour body implants
Dermal fillers
Liposuction body sculpting equipment
Equipements for hair removal (using infra-red, ultraviolet, visible light)
Transcranial simulator
FAQs (Frequently Asked Questions)
Devices with non-medical purposes are classified under which rule?
Apply Annex VIII classification rules in the EU MDR.
Do I need an Authorised Representative for marketing devices without intended medical purpose?
Yes, as per Regulation an AR must be appointed to sell these devices in the Union market.
Have the Common Specifications been released?
No, as of the date Apr 2022 the CS are under the draft stage. Follow our website updates to know more about this or subscribe to our newsletter to get the updates.
SaMD is software intended for one or more medical purposes that perform these without being part of a hardware medical device.
Medical device software is meant to be used, alone or in combination, for a purpose specified in the definition of a “medical device” in the MDR or IVDR, regardless of whether the software is independent or driving or influencing the use of a device.
To be qualified as medical device software, the product must first fulfil the software definition according to this guidance and the description of a medical device according to Article 2(1) of Regulation (EU) 2017/745 – MDR.
To be qualified as an in vitro diagnostic medical device software, the product must additionally fulfil the definition of an in vitro diagnostic medical device according to Article 2(2) of Regulation (EU) 2017/746 – IVDR.
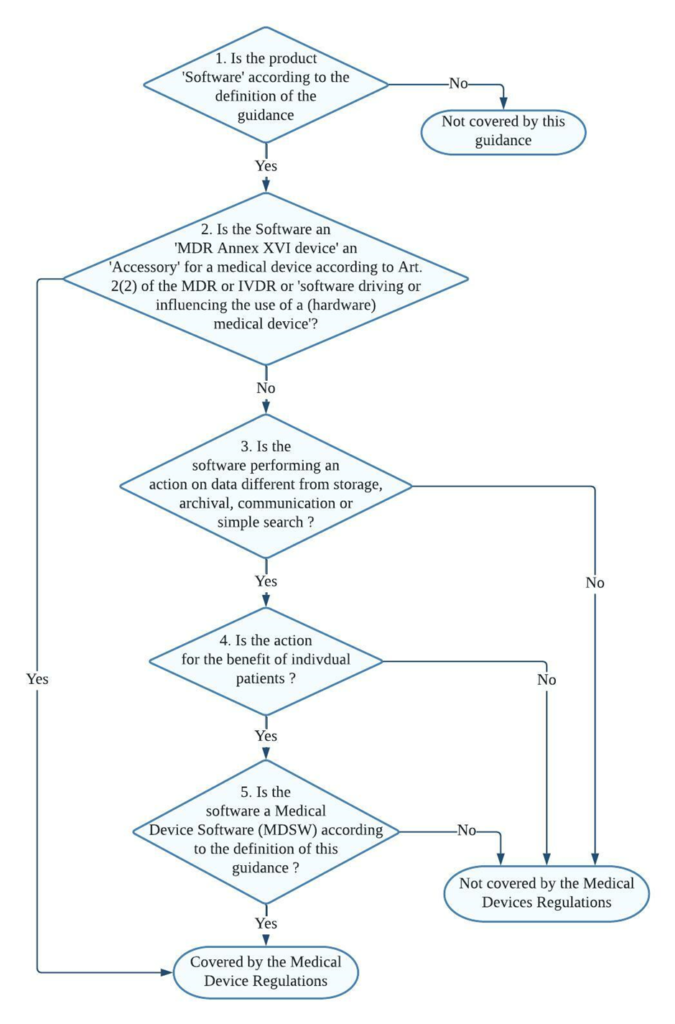
Decision steps for qualification of software as MDSW
Decision step 1: If the product is software according to Section 2 of the guidance (link provided at the bottom), then it may be a medical device software; proceed to decision step 2; else, it is not covered by this guidance but may still be covered by the Medical Devices Regulations.
Decision step 2: If the product is an MDR Annex XVI device or an accessory for a medical device, or is software driving or influencing the use of a medical device, then it must be considered as part of that device in its regulatory process or independently if it is an accessory. If it is not, proceed to decision step 3.
Decision step 3: if the software does perform an action on data or performs an action beyond storage, archival, communication, simple search, lossless compression, then it may be a medical device software; proceed to step 4.
Decision step 4: is the action for the benefit of individual patients?
Examples of software that are not considered as being for the use of individual patients are those which are intended only to aggregate population data, provide generic diagnostic or treatment pathways (not directed to individual patients), scientific literature, medical atlases, models, and templates as well as software intended only for epidemiological studies or registers.
Decision step 5: Is the software medical device software (MDSW) according to the definition of this guidance?
Decision steps for qualification of MDSW as either a medical device or an in vitro diagnostic medical device
Decision Step 1: Does the Medical Device Software (MDSW) provide information within the scope of the in vitro diagnostic medical device definition?
MDSW, which provides information according to Regulation (EU) 2017/746 – IVDR Article 2(2), should qualify as In Vitro Diagnostic Medical Device Software (IVD MDSW)
Concerning a physiological or pathological process or state (by investigation of this process or state)
Concerning congenital physical or mental impairments
Concerning the predisposition to a medical condition or a disease
To determine the safety and compatibility with potential recipients
To predict treatment response or reactions
To define or monitor therapeutic measures.
An MDSW that falls under the definition set out in EU Article 2 (1) of Regulation (EU) 2017/745 – MDR should qualify as Medical Device Software (MD MDSW). In specific, the following considerations should apply to the provision of information by software:
Diagnosis, prevention, monitoring, prediction, prognosis, treatment or alleviation of disease
Diagnosis, monitoring, treatment, alleviation of, or compensation for, an injury or disability
The investigation, replacement, or modification of the anatomy or a physiological or pathological process or state
Control or support of conception
Products specifically intended for the cleaning, disinfection or sterilisation of devices as referred to in Article 1(4) and Annex XVI products.
Decision Step 2: Does the MDSW create information based on data obtained by in vitro diagnostic medical devices only?
Suppose the information provided is based on data obtained solely from in vitro diagnostic medical devices. In that case, the software is an in vitro diagnostic medical device and is, therefore, an IVD MDSW.
If the data analysed is obtained from both in vitro diagnostic and medical devices, proceed to step 3.
Decision Step 3: Is the intended purpose substantially driven by in vitro diagnostic medical devices?
The applicable legislation is Regulation (EU) 2017/746. If the intended purpose is substantially driven by data sources coming from medical devices, then the relevant legislation is Regulation (EU) 2017/745.
In the condition where the intended purpose of the MDSW output data fulfils both the medical device and in vitro diagnostic medical device definitions set out in the MDR and IVDR, a weighting of the data sources based on the significance of the information concerning fulfilling the intended purpose should be conducted to aid the manufacturer in determining which regulation to apply.
Consideration of changes to an MDSW
Manufacturers shall evaluate the potential impact of any changes to the function, intended use, basic design, and manufacturing characteristics on the software’s qualification as MDSW and its classification.
It is to be noted that a change to or the addition of functionality to software may lead it to be qualified as MDSW or a revision of the classification of the MDSW. Similarly, a module that is added to software might be equipped as a MDSW on its own.
When determining the risk class of a combination of a modified MDSW and a medical device, the intended purpose and functionality of that (new) combination must be considered.
FAQs
How MDD considered SaMD classification approach and what difference has MDR brought in it ?
Most standalone medical software falls into the lowest risk category — Class I — under the EU MDD. The only software used for specialised diagnostic activities obtained a higher classification.
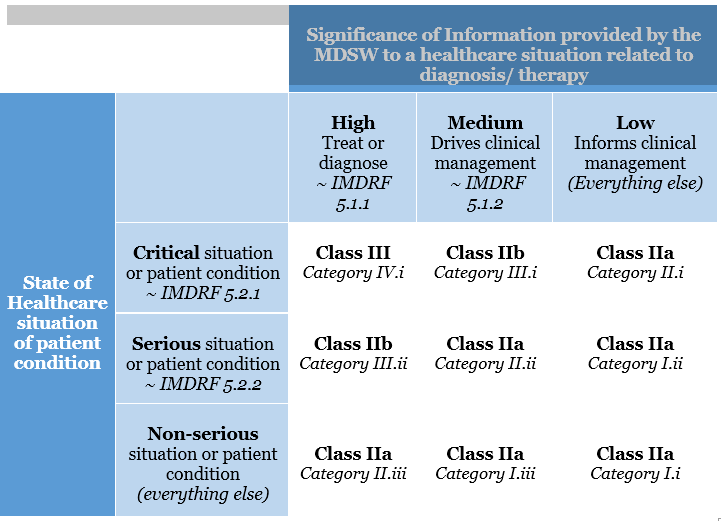
This categorisation has been altered in the EU MDR. The risk classification of software is covered under Rule 11. The legislation specifies that software used for diagnostic or therapeutic purposes must be classified as Class IIa (or higher). Rule 11 was added to the MDR to address the risks associated with information provided by an active device, such as the MDSW. The significance of the information provided by the active device to the healthcare decision (patient management) in connection with the healthcare situation is described and categorised in Rule 11.
Rule 11 states:
Software intended to provide information that is used to make decisions with diagnosis or therapeutic purposes is classified as class IIa, except if such decisions have an impact that may cause:
Death or an irreversible deterioration of a person’s state of health, in which case it is in class III
Severe deterioration of a person’s state of health or surgical intervention, in which case it is classified as class IIb.
Software intended to monitor physiological processes is classified as class IIa, except if it is designed for monitoring vital physiological parameters, where the nature of variations of those parameters is such that it could result in immediate danger to the patient, in which case it is classified as class IIb.
All other software is classified as class I.
What is the transition plan from MDD to MDR for SaMD devices ?
While many devices approved under the MDD can continue to be sold until 2024, Class I items and those that have been “up-classified” due to the MDR do not have that opportunity; they must be compliant by May 2020.
The MDR regulation’s wording has been clarified – it’s all about the expiration dates of CE mark certificates. MDD-certified products can be used until the expiration date on their CE-marking certificate (a soft transition, potentially up to 2024). However, there are specific additional requirements, such as post-market surveillance. On the other hand, Class I products are self-certified and so lack a certificate with an expiration date. At the end of the transition period, they must be compliant on May 26, 2020.
How is MDSW classified under IVDR ?
In determining the proper classification of MDSW under the IVDR, the manufacturer shall consider all categories and implement the rules of Annex VIII of the IVD Regulation (EU) 2017/746.
Examples for the classification of MDSW under the IVDR:
Software intended to be installed on a fully automated enzyme-linked immunosorbent assay (ELISA) analyser, and intended to determine the Human HbA1c concentration in serum from the results obtained with a Human HbA1c ELISA, designed to screen for and diagnose diabetes and monitor diabetic patients, should be in class C per Rule 3(k).
Software within a PAP stain automated cervical cytology screening system intended to classify the PAP cervical smear as usual or suspicious should be in class C per Rule 3(h).
Software for the interpretation of automated readings of line immunoassay to confirm and determine antibodies to HIV-1, HIV-1 group O and HIV-2 in human serum and plasma should be in class D per Rule 1.
Software that uses maternal parameters such as age, the concentration of serum markers and information obtained through foetal ultrasound examination for evaluating the risk of trisomy 21 should be in class C per Rule 3(l).
Classification examples in Annex IV are provided for guidance purposes and illustrate how a particular rule may be applied to a device. The indicated classification in the example is not a confirmation of the final classification of the device, as other laws must also be considered.
What are the risks SaMD medical devices possess ?
While SaMD has the potential to improve the healthcare system significantly, the International Medical Device Regulators Forum (IMDRF), a collection of medical device regulators working toward international regulatory harmonisation, has recognised that SaMD poses new problems to medical device regulation.
Regulators face the difficult task of regulating software that undergoes frequent changes, potentially affecting the software’s safety and efficacy. Because the conventional physical constraints of containment are no longer present, the ability to download the software through the internet creates a risk.
To identify and track SaMD throughout its life, regulators and the industry must agree on globally applicable device identification and coding standard. They’re not like a standard medical device, which can be labelled with a Unique Device Identifier on the outside.
Another issue with SaMD is the software’s security. The medical device’s use may be jeopardised due to cybersecurity vulnerabilities. It could allow the attacker to take control of the device remotely, modify its operation and compromise its safety or effectiveness, or expose confidential data.
From a regulatory standpoint, the following are some of SaMD’s challenges:
Medical Device Regulations establish guidelines for categorising medical devices from low to high risk. Some SaMD do not fall neatly into the classification scheme designed for traditional medical devices.
Artificial Intelligence is already improving several SaMD systems in image-based healthcare by continuously learning from the data provided to the software.
MSDW Risk Classification based on Rule 11 of the MDR
Any action performed to reduce the risk of death or serious deterioration in health connected with the use of a medical device is referred to as Field Safety Corrective Action (FSCA). The manufacturer is required to take action to remove or limit the risk of the recognised dangers.
If a medical device malfunctions in Switzerland, the manufacturer is required to undertake an FSCA and Swissmedic keeps track of all FSCAs regarding medical equipment sold in Switzerland.
Reporting a Field Safety Corrective Action (FSCA)
Field safety corrective actions involving items placed on the Swiss market must be reported to Swissmedic by manufacturers
The Swiss authorised representative is responsible for reporting for manufacturers who are not headquartered in Switzerland
When a field safety corrective action is recorded, Swissmedic determines whether the risk can be effectively mitigated by the manufacturer’s steps and supervises their execution
The content is the responsibility of the manufacturer or an authorised representative (accuracy, completeness, and data protection)
Importers must immediately notify the manufacturer and its Swiss authorised agent of any complaints or reports of suspected incidents involving a device they have placed on the market
Distributors who receive complaints or reports about a device they sold must immediately notify the manufacturer, as well as the manufacturer’s Swiss authorised agent and the importer, if relevant
Healthcare professionals also must report serious occurrences to the supplier and Swissmedic
Such reports should be sent as soon as possible to the supplier organization with the help of the form available on the Swissmedic website.
A combined Initial-Final Report (combined initial-final MIR)
Evaluation by Swissmedic
Electronic submission
FSCAs must be reported with the help of the form released by Swissmedic. The Swissmedic website has this form available for download. This form must be used to send all FSCA reports to Swissmedic in an electronic, machine-readable manner.
Reports can be submitted in English or one of the official Swiss languages. All required fields must be filled out. The completed report form, the field safety notice (FSN), the customer list, and any other supporting paperwork should be emailed to [email protected].
If Swissmedic has any additional questions about an FSCA, it will contact you by email.
Timeline for reporting
If the serious incident clearly poses or has the potential to pose, a serious and imminent threat to the lives or health of a large number of people (serious public health threat), the report must be submitted as soon as possible, and no later than 2 calendar days after becoming aware of the incident
If the significant incident resulted in death or an unexpected serious worsening in a person’s health, the report must be submitted immediately, and at the latest within 10 calendar days
All other major incidents must be reported as soon as possible and no later than 15 calendar days after the date of discovery
An Initial Report must be made to Swissmedic if the manufacturer or Swiss authorised representative has not received sufficient information within the statutory time limit to determine whether or not a reportable occurrence has occurred
Types of Reports
1. Reporting of Trends
If a manufacturer sees a statistically significant increase in the frequency or severity of non-serious occurrences or expected negative side effects, they must notify SwissMedic by sending the Trends report form to mailto:[email protected].
The Swiss authorised representative is in charge of this role for producers who are not headquartered in Switzerland. The manufacturer or the Swiss approved representative might submit the report in this scenario.
2. Periodic Summary Report (PSR)
Serious incidents where the root cause is known (and/or) the serious incident is already the subject of an FSCA (and/or) the serious incidents occur frequently and are well documented then these incidents can be grouped and reported together to SwissMedic in the form of Periodic summary Report.
This process can be initiated by sending SwissMedic an email of this Periodic summary report form in English or any of the Swiss national languages.
3. Periodic Safety Update Report (PSUR)
Manufacturers of class IIa, IIb, and III medical devices must develop and submit a Periodic Safety Update Report (PSUR) for each product and, if relevant, for each product category or group, to the responsible notified body.
The PSUR and the Notified Body’s evaluation outcome must be submitted to Swissmedic upon request by the manufacturer or its Swiss authorised agent.
FAQ:
What is Field Safety Notice?
Every manufacturer is required to inform its users and, if applicable, patients about field safety remedial actions that have been implemented. Typically, this involves sending a Field Safety Notice (FSN).
The European Commission website has FSN templates as well as a confirmation form. The templates are designed to help manufacturers create high-quality customer letters that include all relevant information.
In the event of publishing, the FSN must not contain any information that would be in violation of data protection laws. Before publication, particularly sensitive personal data should be removed or, if absolutely necessary, anonymized.
Do Hospitals have to maintain a medical device vigilance system?
Yes, Hospitals are legally required to establish a reporting system. They must designate someone to be responsible for serious occurrences involving medical devices and they must publicly notify Swissmedic of this vigilant contact person for medical devices.
If there are any new vigilance contact persons for medical devices, or if the contact details of existing registered contact persons change, Swissmedic must be notified. Swissmedic inspects the reporting systems in hospitals.