SaMD is software intended for one or more medical purposes that perform these without being part of a hardware medical device.
Medical device software is meant to be used, alone or in combination, for a purpose specified in the definition of a “medical device” in the MDR or IVDR, regardless of whether the software is independent or driving or influencing the use of a device.
You can read more on the SaMD Regulation here.
To be qualified as medical device software, the product must first fulfil the software definition according to this guidance and the description of a medical device according to Article 2(1) of Regulation (EU) 2017/745 – MDR.
To be qualified as an in vitro diagnostic medical device software, the product must additionally fulfil the definition of an in vitro diagnostic medical device according to Article 2(2) of Regulation (EU) 2017/746 – IVDR.
SaMD Devices Classification Steps
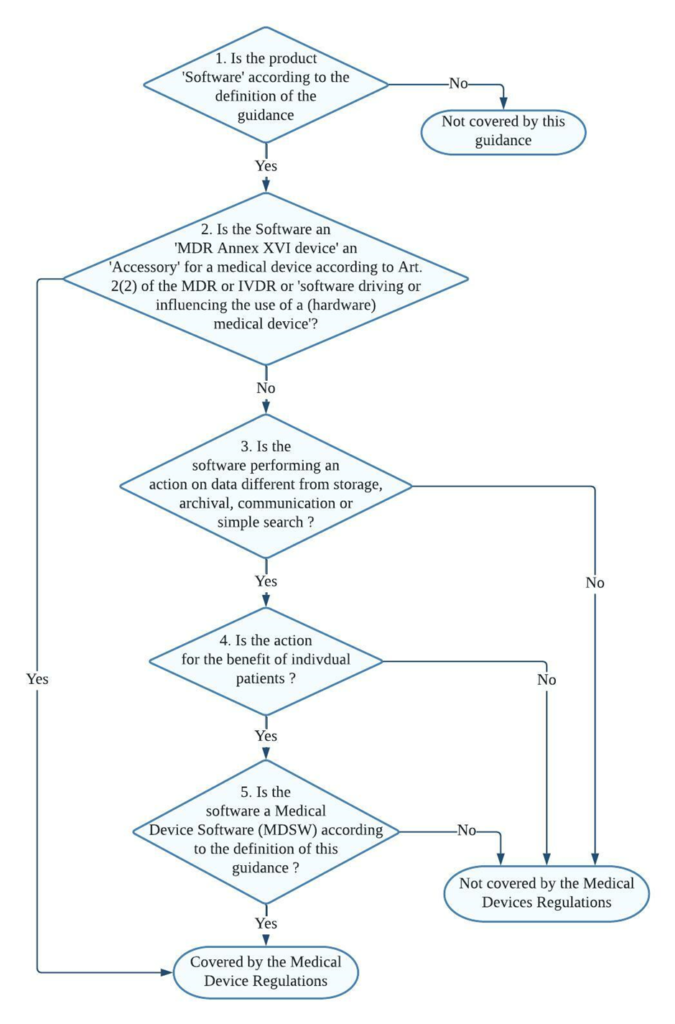
Decision steps for qualification of software as MDSW
Decision step 1: If the product is software according to Section 2 of the guidance (link provided at the bottom), then it may be a medical device software; proceed to decision step 2; else, it is not covered by this guidance but may still be covered by the Medical Devices Regulations.
Decision step 2: If the product is an MDR Annex XVI device or an accessory for a medical device, or is software driving or influencing the use of a medical device, then it must be considered as part of that device in its regulatory process or independently if it is an accessory. If it is not, proceed to decision step 3.
Decision step 3: if the software does perform an action on data or performs an action beyond storage, archival, communication, simple search, lossless compression, then it may be a medical device software; proceed to step 4.
Decision step 4: is the action for the benefit of individual patients?
Examples of software that are not considered as being for the use of individual patients are those which are intended only to aggregate population data, provide generic diagnostic or treatment pathways (not directed to individual patients), scientific literature, medical atlases, models, and templates as well as software intended only for epidemiological studies or registers.
Decision step 5: Is the software medical device software (MDSW) according to the definition of this guidance?
Decision steps for Qualification of MDSW as either a Medical Device or an In Vitro Diagnostic Medical Device
Decision Step 1: Does the Medical Device Software (MDSW) provide information within the scope of the in vitro diagnostic medical device definition?
MDSW, which provides information according to Regulation (EU) 2017/746 – IVDR Article 2(2), should qualify as In Vitro Diagnostic Medical Device Software (IVD MDSW)
1. Concerning a physiological or pathological process or state (by investigation of this process or state)
2. Concerning congenital physical or mental impairments
3. Concerning the predisposition to a medical condition or a disease
4. To determine the safety and compatibility with potential recipients
5. To predict treatment response or reactions
6. To define or monitor therapeutic measures.
An MDSW that falls under the definition set out in EU Article 2 (1) of Regulation (EU) 2017/745 – MDR should qualify as Medical Device Software (MD MDSW). In specific, the following considerations should apply to the provision of information by software:
1. Diagnosis, prevention, monitoring, prediction, prognosis, treatment or alleviation of disease
2. Diagnosis, monitoring, treatment, alleviation of, or compensation for, an injury or disability
3. The investigation, replacement, or modification of the anatomy or a physiological or pathological process or state
4. Control or support of conception
5. Products specifically intended for the cleaning, disinfection or sterilisation of devices as referred to in Article 1(4) and Annex XVI products.
Decision Step 2: Does the MDSW create information based on data obtained by in vitro diagnostic medical devices only?
Suppose the information provided is based on data obtained solely from in vitro diagnostic medical devices. In that case, the software is an in vitro diagnostic medical device and is, therefore, an IVD MDSW.
If the data analysed is obtained from both in vitro diagnostic and medical devices, proceed to step 3.
Decision Step 3: Is the intended purpose substantially driven by in vitro diagnostic medical devices?
The applicable legislation is Regulation (EU) 2017/746. If the intended purpose is substantially driven by data sources coming from medical devices, then the relevant legislation is Regulation (EU) 2017/745.
In the condition where the intended purpose of the MDSW output data fulfils both the medical device and in vitro diagnostic medical device definitions set out in the MDR and IVDR, a weighting of the data sources based on the significance of the information concerning fulfilling the intended purpose should be conducted to aid the manufacturer in determining which regulation to apply.
Consideration of changes to an MDSW
Manufacturers shall evaluate the potential impact of any changes to the function, intended use, basic design, and manufacturing characteristics on the software’s qualification as MDSW and its classification.
It is to be noted that a change to or the addition of functionality to software may lead it to be qualified as MDSW or a revision of the classification of the MDSW. Similarly, a module that is added to software might be equipped as a MDSW on its own.
When determining the risk class of a combination of a modified MDSW and a medical device, the intended purpose and functionality of that (new) combination must be considered.
FAQs
How MDD considered SaMD classification approach and what difference has MDR brought in it ?
Most standalone medical software falls into the lowest risk category — Class I — under the EU MDD. The only software used for specialised diagnostic activities obtained a higher classification.
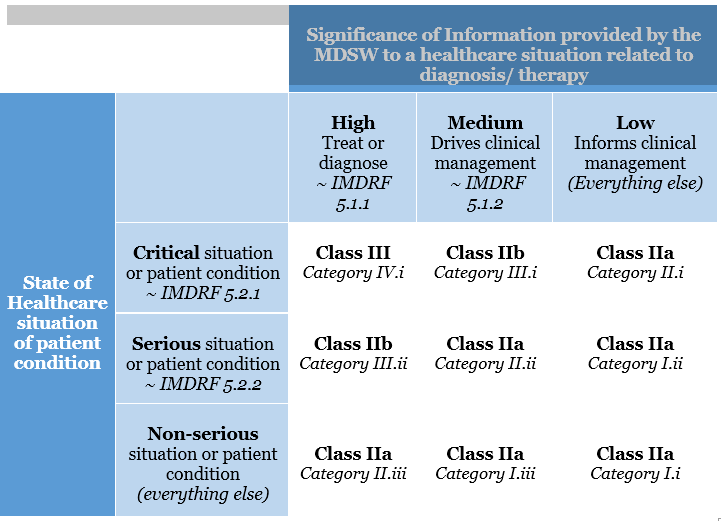
This categorisation has been altered in the EU MDR. The risk classification of software is covered under Rule 11. The legislation specifies that software used for diagnostic or therapeutic purposes must be classified as Class IIa (or higher). Rule 11 was added to the MDR to address the risks associated with information provided by an active device, such as the MDSW.
The significance of the information provided by the active device to the healthcare decision (patient management) in connection with the healthcare situation is described and categorised in Rule 11.
Rule 11 states:
Software intended to provide information that is used to make decisions with diagnosis or therapeutic purposes is classified as class IIa, except if such decisions have an impact that may cause:
1. Death or an irreversible deterioration of a person’s state of health, in which case it is in class III
2. Severe deterioration of a person’s state of health or surgical intervention, in which case it is classified as class IIb.
Software intended to monitor physiological processes is classified as class IIa, except if it is designed for monitoring vital physiological parameters, where the nature of variations of those parameters is such that it could result in immediate danger to the patient, in which case it is classified as class IIb.
All other software is classified as class I.
What is the transition plan from MDD to MDR for SaMD devices ?
While many devices approved under the MDD can continue to be sold until 2024, Class I items and those that have been “up-classified” due to the MDR do not have that opportunity; they must be compliant by May 2020.
The MDR regulation’s wording has been clarified – it’s all about the expiration dates of CE mark certificates. MDD-certified products can be used until the expiration date on their CE-marking certificate (a soft transition, potentially up to 2024). However, there are specific additional requirements, such as post-market surveillance. On the other hand, Class I products are self-certified and so lack a certificate with an expiration date. At the end of the transition period, they must be compliant on May 26, 2020.
How is MDSW classified under IVDR ?
In determining the proper classification of MDSW under the IVDR, the manufacturer shall consider all categories and implement the rules of Annex VIII of the IVD Regulation (EU) 2017/746.
Examples for the classification of MDSW under the IVDR:
1. Software intended to be installed on a fully automated enzyme-linked immunosorbent assay (ELISA) analyzer, and intended to determine the Human HbA1c concentration in serum from the results obtained with a Human HbA1c ELISA, designed to screen for and diagnose diabetes and monitor diabetic patients, should be in class C per Rule 3(k).
2. Software within a PAP stain automated cervical cytology screening system intended to classify the PAP cervical smear as usual or suspicious should be in class C per Rule 3(h).
3. Software for the interpretation of automated readings of line immunoassay to confirm and determine antibodies to HIV-1, HIV-1 group O and HIV-2 in human serum and plasma should be in class D per Rule.
4. Software that uses maternal parameters such as age, the concentration of serum markers and information obtained through foetal ultrasound examination for evaluating the risk of trisomy 21 should be in class C per Rule 3(l).
Classification examples in Annex IV are provided for guidance purposes and illustrate how a particular rule may be applied to a device. The indicated classification in the example is not a confirmation of the final classification of the device, as other laws must also be considered.
What are the risks SaMD medical devices possess ?
While SaMD has the potential to improve the healthcare system significantly, the International Medical Device Regulators Forum (IMDRF), a collection of medical device regulators working toward international regulatory harmonisation, has recognised that SaMD poses new problems to medical device regulation.
Regulators face the difficult task of regulating software that undergoes frequent changes, potentially affecting the software’s safety and efficacy. Because the conventional physical constraints of containment are no longer present, the ability to download the software through the internet creates a risk.
To identify and track SaMD throughout its life, regulators and the industry must agree on globally applicable device identification and coding standard. They’re not like a standard medical device, which can be labelled with a Unique Device Identifier on the outside.
Another issue with SaMD is the software’s security. The medical device’s use may be jeopardised due to cybersecurity vulnerabilities. It could allow the attacker to take control of the device remotely, modify its operation and compromise its safety or effectiveness, or expose confidential data.
From a regulatory standpoint, the following are some of SaMD’s challenges:
1. Medical Device Regulations establish guidelines for categorising medical devices from low to high risk. Some SaMD do not fall neatly into the classification scheme designed for traditional medical devices.
2. Artificial Intelligence is already improving several SaMD systems in image-based healthcare by continuously learning from the data provided to the software.
MSDW Risk Classification based on Rule 11 of the MDR