

Blog
Trending Now

EU Publishes New Regulation to Address In Vitro Diagnostic Device (IVDD) Shortages: Key Updates and Recommendations for Manufacturers
On July 9, 2024, Regulation (EU) 2024/1860 was published in the Official Journal of the European Union (OJEU) and took effect immediately. This new regulation aims to address the...

September Newsletter
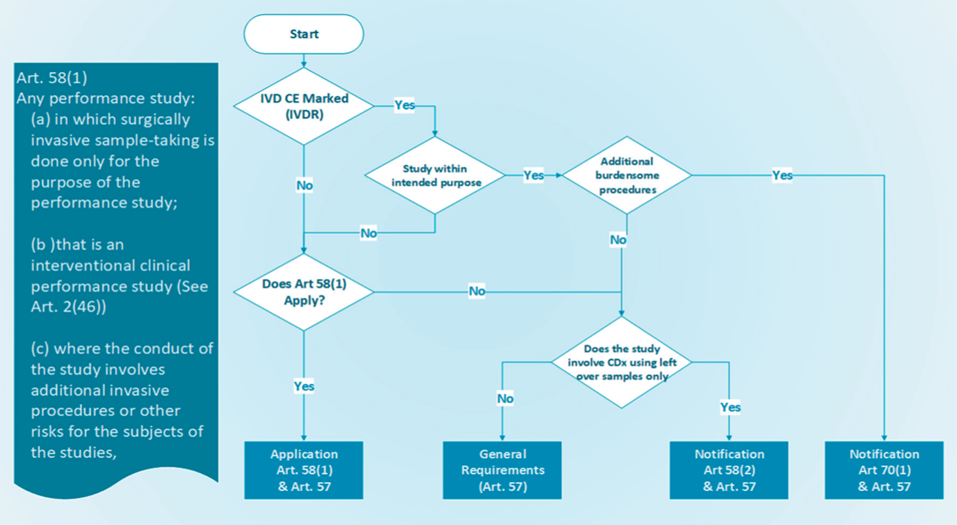
EU REGULATIONS The expert panel in the field of in vitro diagnostic medical devices now accepts submissions from notified bodies for the Performance Evaluation Consultation Pro...
October Newsletter
EU REGULATIONS The expert panel in the field of in vitro diagnostic medical devices now accepts submissions from notified bodies for the Performance Evaluation Consultation Pro...
November Newsletter
EUROPE EUDAMED Notified Bodies and Certificates Module – Relevant documents and information| 15 November 2021 Regulation (EU) 2017/745 on medical devices (MDR) and Re...
December Newsletter
EU REGULATIONS MDCG 2022-4 – Guidance on appropriate surveillance regarding the transitional provisions under Article 120 of the MDR | 16 February 2022 Article 120(2) and...
January Newsletter 2022
January Newsletter – EU Regulations MDCG 2022-1: Notice to 3rd country manufacturers of SARS-CoV-2 In vitro diagnostic medical devices | 12 January 2022 This not...
February Newsletter 2022
February Newsletter: EU REGULATIONS MDCG 2022-4 – Guidance on appropriate surveillance regarding the transitional provisions under Article 120 of the MDR | 16 February 2...
March Newsletter
AUSTRALIA March Newsletter: Consumer/Patient information materials requirements | 02 March 2022 On 26 October 2017, the Australian government passed laws requiring impl...
April Newsletter
AUSTRALIA Transitioning to the new Therapeutic Goods Advertising Code | 05 April 2022 The Therapeutic Goods Administration (TGA) is updating advertisers on the transitional...
May Newsletter 2022
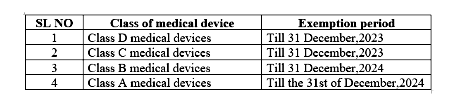
EUROPEAN UNION (EU) Stricter rules for placing medical tests on the market | 25 May 2022 From 26 May 2022, new laws for in vitro diagnostic medical devices (IVDR) such as HI...
June Newsletter
EUROPEAN UNION (EU) - JUNE NEWSLETTER Guidance document on the implementation of EU Product Rules 2022 | 29 June 2022 The ‘Blue Guide’ on EU product rules 2022 (Document...
July Newsletter 2022
EUROPEAN UNION (EU) Harmonised administrative practices and alternative technical solutions| 13 July 2022 The MDCG guidance document advises the Member States and other rele...
August Newsletter 2022
EUROPEAN UNION (EU) MDCG guidance on Notified body capacity and availability of medical devices and IVDs |26 August 2022 The MDCG acknowledges that there are still significa...
September Newsletter 2022
EUROPEAN UNION (EU) Notified Bodies Position paper on application of hybrid audits for QMS assessments under MDR and IVDR|26 September 2022 The latest position paper discuss...
October Newsletter 2022
Guidance on EU Authorized Representatives | 31 October 2022 Medical Device Coordination Group has updated its guidance on Authorized Representatives under MDR 2017/745 and IVDR...
November Newsletter
EUROPEAN UNION (EU) EU guidance on reference laboratories for IVD medical devices | 30 November 2022 EU Commission has published new guidance for candidate reference laborat...
February Newsletter
EUROPEAN UNION (EU) MDCG guidance on vigilance terms and concepts of EU MDR | 14 February 2023 The MDCG 2023-3 guidance document clarifies some of the essential terms and co...
March Newsletter 2023
EUROPEAN UNION (EU) Guidance on MDR transitional period extension | 28 March 2023 The amendment of the MDR and of the IVDR through Regulation (EU) 2023/607 aims to ensure a...
April Newsletter 2023
Germany Database for In vitro diagnostics Medical Devices notification | 25 April 2023 In Vitro Diagnostic Medical Devices Notifications includes notifications on the first...
May Newsletter 2023
EUROPEAN UNION (EU) Updated MHRA guidance on significant changes under MDR | 12 May 2023 The guidance document MDCG 2020-3 Rev.1 is intended to clarify the concept of ‘sig...
Regulations updates






Five minute reads
")
Cosmetic Regulatory Requirements in Sweden
In Sweden, cosmetic products are regulated primarily under the framework of the European Union (EU) legislation, specifically Regulation (EC) No 1223/2009 on cosmetic products. This regulation ensures the safety and compliance of...
")
")
")
")
")
Five Minute Reads




Cosmetic Blogs
")
In Sweden, cosmetic products are regulated primarily under the framework of the European Union (EU) legislation, specifically Regulation (EC) No 1223/2009 on cosmetic products. This regulation ensures the safe
")
In the United Kingdom (UK), cosmetic products are regulated under the UK Cosmetics Regulation 2019, which governs the safety and marketing of cosmetic products. This regulation largely mirrors the European Uni
")
Cosmetic Regulatory Requirements in Belgium follows the EU regulation (Regulation (EC) No 1223/2009) without additional national requirements for cosmetics. Federal Agency for Medicines and Health Products
")
Cosmetic Regulatory Requirements in France are regulated under European Union (EU) legislation, specifically Regulation (EC) No 1223/2009 on cosmetic products. The French National Agency for Medicines and Heal
")
Cosmetic Regulatory Requirements in Norway are regulated under the framework of the European Economic Area (EEA) agreement, which aligns Norwegian regulations closely with those of the European Union (EU). The
 (1)")
Spain follows the European Union (EU) regulations for cosmetics, primarily governed by Regulation (EC) No 1223/2009. This regulation ensures the safety and compliance of cosmetic products marketed within the E
")
Cosmetic Regulatory requirements in Denmark adheres to the EU regulation (Regulation (EC) No 1223/2009). The Danish Environmental Protection Agency (EPA) is the responsible authority for cosmetic regulations a
")
Italy adheres to the EU regulation (Regulation (EC) No 1223/2009). The Italian Ministry of Health oversees the regulation of cosmetic products in the country. Italian Ministry of Health In Italy, the ove
")
Poland regulates cosmetic products under the framework of the European Union (EU) legislation, specifically adhering to Regulation (EC) No 1223/2009 on cosmetic products. This regulation ensures the safety and

Understanding the distinctions between product classes within the European cosmetics landscape can be complex and ambiguous, often requiring input from authoritative bodies. Certain products may share simi

In recent years, the beauty and cosmetics industry has witnessed exponential growth worldwide, breaking down geographical barriers and creating a global marketplace. One country that has embraced this trans

eCTD-an overview In today’s economy, life science companies are seeking for strategies to continue growing despite impending patent expirations, increasing generic competition, and rising drug development

The term “animal testing” refers to procedures carried out on living animals for research into basic biology and diseases, evaluating the efficacy of new pharmaceuticals, and testing the safety of consumer

Cosmetic ingredients and products are not required to have FDA clearance before being sold in the U.S. The only exception is colour additives, which need to be authorised for their intended purpose (apart from

Title 21 of the Code of Federal Regulations (CFR), section 7.3(g), describes a recall as a firm’s removal or correction of a marketed product that FDA judges to be in violation of the laws we administer and

by OMC Medical | Dec 13, 2023 | Cosmetics, EU Understanding the distinctions between product classes within the European cosmetics landscape can be complex and ambiguous, often requiring input from au
Pharma Blogs
")
Why South Korea is a Booming Market for Medical Devices?
South Korea has emerged as a significant player in the global medical device market, and its growth trajectory is impressive. The convergence of government support, advanced technology, a strong domestic market, and strategic tra...
")




Pharma Blogs

TFDA Requirements for Summary Product Characteristic (SPC) Name of the Medical Product&nbs

The Indonesian Food and Drug Authority (BPOM) plays a crucial role in ensuring the safety and effic

Territory: Southeast Asia Official Language: Indonesian/English Regulat

MoH: Armenia Ministry of Health MoH website: https://www.gov.am/en/structure/1/ Reg

MoH: Ministry of Health of Argentina MoH website: https://www.argentina.gob.ar/salud

MoH: The Ministry of Health and Social Protection. MoH website: https://shendetesia.go

MoH: Ministry of Public Health MoH website: https://moph.gov.af/en Regulatory Autho

A medical device’s intended use and inherent risks must be considered when determining its MDR cl

Technical documentation should contain details of the medical device in a clear, organized, readily

“Professional use” Test Kit – Procedure for registration in UK Novel Corona Virus Di


Labelling Requirements – Swiss Authorized Representative Written, printed, or graphic informat

Ministry of Health: Ministry of Health Regulatory Authority:&nb

A medical device’s intended use and inherent risks must be considered when determining its MDR cl

Navigating Medical Device Registration with Iraq’s Regulatory Authority: KIMADIA Guidelines and R

“Professional use” Test Kit – Procedure for registration in UK Novel Corona Virus Di

South Korea has emerged as a significant player in the global medical device market, and its growth
Medical Device and IVD Blogs

Switzerland Authorised Representative
Medical devices are regulated by the Swiss Agency for Therapeutic Product Manufacturer Responsibility 1.Ensuring medical devices are placed on the market is in accordance with the requirement of the

Switzerland Medical Device Regulations
Switzerland Market Situation As the Institutional Agreement is missing (InstA) is missing, the EU has not updated the Mutual Recognition Agreement (MRA) No barrier-free access to the EU internal market

Swiss Regulation QA WHITE PAPER
MEDICAL DEVICE REGULATION UPDATE IN SWITZERLAND 1. Who is responsible for medical device regulation in Switzerland? The Swiss Agency regulates medical devices for Therapeutic Products (Swiss medic) 2.

Switzerland AR Responsibilities
Obligations of the Switzerland AR Responsibilities 1. Verify the DoC and technical documentation have been drawn up and where necessary conformity assessment procedure has been carried out. 2. Keep avail

Swiss Manufacturer Obligations
1. Manufacturers outside Switzerland will need to appoint Swiss AR. 2. The Manufacturer will use commercially reasonable efforts to update its technicaldocumentation for the devices, to comply with the requ

Labelling Requirements – Swiss Authorized Representative
Labelling Requirements – Swiss Authorized Representative Written, printed, or graphic information appears either on the device itself or on the packaging of each unit or the packaging of multiple devices

Nomenclature of Medical Devices
Medical device nomenclatures are those products used to prevent, diagnose, treat, and monitor the many diseases known to humankind. Medical devices and medicines play an equally important role in treating huma
