
EUROPEAN UNION (EU)
Stricter rules for placing medical tests on the market | 25 May 2022
From 26 May 2022, new laws for in vitro diagnostic medical devices (IVDR) such as HIV testing, pregnancy tests, and COVID-19 tests are in effect. The guidelines will improve public health and patient safety for these devices, putting EU law in line with technological advancements and medical research progress. It also ensures equitable market access for manufacturers by unifying market access standards across the EU Member States.
In summary, the In Vitro Diagnostic Medical Devices Regulation introduces three significant advancements:
- In vitro diagnostic medical equipment’s quality, safety, and reliability are improved.
- Transparency and information for patients are improved.
- Improves market surveillance and vigilance
Interface between Regulation (EU) 536/2014 on clinical trials for medicinal products for human use (CTR) and Regulation (EU) 2017/746 on in vitro diagnostic medical devices (IVDR) | 25 May 2022
The Q&A document aims to clarify some of the overlaps between the Clinical Trials for Medicinal Products for Human Use (CTR) and the In Vitro Diagnostic Medical Devices Regulation (EU) 2017/746 (IVDR). Clinical trial experts from the Clinical Trials Facilitation and Coordination Group (CTFG) and in vitro diagnostics experts from the Medical Device Coordination Group (MDCG) collaborated on it. Clinical trial assays can range from CE-marked in vitro diagnostic medical devices (IVDs) to trial- or medicinal product-specific assays that aren’t necessarily aimed at becoming IVDs. This document and the idea of the “medical purpose of an assay in a clinical trial” as a specific context were born out of the necessity to define criteria for these tests.
Status of the EU-Switzerland Mutual Recognition Agreement (MRA) for in vitro diagnostic medical devices | 24 May 2022
Switzerland has participated in the European Union’s internal market for in vitro diagnostic medical devices through the EU-Switzerland Mutual Recognition Agreement’s medical devices chapter (MRA). The MRA’s medical devices chapter allows the European Union and Switzerland to recognise conformity assessment certificates based on the equivalence of Directive 98/79/EC on in vitro diagnostic medical devices and Swiss legislation. The parties have been able to trade in vitro diagnostic medical equipment more quickly.
On 26 May 2022, the new Regulation (EU) 2017/746 on in vitro diagnostic medical devices replaced Directive 98/79/EC. In the absence of an MRA update to incorporate Regulation (EU) 2017/746, the part of the MRA chapter covering in vitro diagnostic medical devices ceases to apply as of 26 May 2022.
Application of IVDR requirements to ‘legacy devices’ and to devices placed on the market before 26 May 2022 | 20 May 2022
The document provides information on the application of IVDR regulations to ‘legacy devices’ and ‘old’ devices since Regulation (EU) 2022/1121 extended the transitional provisions of Regulation (EU) 2017/746 on in vitro diagnostic medical devices (IVDR), specifically Article 110(3).
Unique Device Identification system under Regulation (EU) 2017/745 and Regulation (EU) 2017/746 | 20 May 2022
The Q&A documentcovers the concerns about the Unique Device Identification system (UDI system), which was established by Regulation (EU) 2017/745 on medical devices (MDR) and Regulation (EU) 2017/746 on in vitro diagnostic medical devices (IVDR). The document intends to give economic operators more information on applying and implementing the UDI requirements.
Summary of Safety and Performance template | 20 May 2022
For class C and D devices, other than devices for performance studies, IVD Regulation (EU) 2017/746 requires the manufacturer to prepare a Summary of Safety and Performance (SSP), which provides public access to an up-to-date summary of the main aspects of the safety and performance of the device. A notified body (NB) must confirm the SSP before it may be made public via the European database on medical devices (EUDAMED). The template can be found here.
New publication of Harmonised standards under the medical devices Regulations | 17 May 2022
The European Commission issued a decision on 11 May 2022, amending the relationship between harmonised criteria for quality management systems, sterilisation, and risk management applications in medical devices. The annex to implementing decision is amended as follows:
- Entry 10 is replaced by EN ISO 13485:2016 – Medical devices – Quality management systems – Requirements for regulatory purposes (ISO 13485:2016)
- EN ISO 13485:2016/AC:2018, EN ISO 13485:2016/A11:2021
- The following standards have been added:
- EN 285:2015+A1:2021 – Sterilisation – Steam sterilisers – Large sterilisers
- EN ISO 14971:2019 – Medical devices – Application of risk management to medical devices (ISO 14971:2019)
- EN ISO 14971:2019/A11:2021
Significant changes regarding the transitional provision under Article 110(3) of the IVDR | 04 May 2022
This guidance document aims to clarify the idea of ‘significant changes in the design and intended purpose’ as defined by IVDR Article 110. (3). It applies to manufacturers of devices that comply with Directive 98/79/EC and the devices that are placed on the market or put into operation after 26 May 2022 during the transition period, in line with Article 110(3) IVDR, regardless of whether those devices needed notified body involvement under the IVDD.
AUSTRIA
Clinical investigations with medical devices | 25 May 2022
With the implementation of IVD Regulation (EU) 2017/746, which will regulate the performance studies of in vitro diagnostic medical devices, from 26 May 2022, performance studies must therefore be submitted or reported to the BASG for approval using the updated form, following the requirements of the IVDR. For clinical investigations, the valid submission will then be confirmed. The study can be initiated, followed by a 45-day scientific and regulatory review period.
IRELAND
In Vitro Diagnostic Medical Devices Regulation comes into effect in the EU | 26 May 2022
The European Union’s In Vitro Diagnostic Medical Devices Regulation (IVDR) 2017/746 took effect on 26 May 2022. IVDs are medical devices that use biological samples to do tests to evaluate a person’s health condition. They range from self-tests for pregnancy and blood glucose testing for people with diabetes to complex diagnoses performed in clinical laboratories.
IVDR Transition Timelines | 26 May 2022
The EU Medical Devices Directive 98/79/EC (IVDD) will be replaced by Regulation (EU) 2017/746 on in vitro diagnostic medical devices (IVDR), which took effect on 26 May 2022. The existing transitional timeframes for devices set out in Article 113 of the IVDR are extended by this amendment. The IVDR classification will determine the length of the transition. By 26 May 2022, self-declared IVDs that will not be classified under the IVDR must be IVDR compliant. Below are the transition timelines:
| IVDR Classification | It may be placed on the market until | Can continue to be made available until |
| Class D | 26 May 2025 | 26 May 2026 |
| Class C | 26 May 2026 | 26 May 2027 |
| Class B | 26 May 2027 | 26 May 2028 |
| Class A (sterile) | 26 May 2027 | 26 May 2028 |
In-House Manufactured Devices (IHM) – IVDR Article 5 | 26 May 2022
Article 5(5) of the IVDR outlines the precise criteria and duties that apply to health institutions that manufacture and use IVDs in-house, as long as the device is not transferred to another legal entity. From 26 May 2022, IHM devices will be subject to the general safety and performance criteria outlined in Annex I of the IVDR. However, Regulation 2022/112 defers several IHM device requirements, which are listed in the table below:
| IVDR requirements | Description | Applicable From |
| Article 5(5) and Annex I | Annex I applies to in house manufactured devices. | 26 May 2022 |
| Article 5(5) b, c, and e-i | See Art 5(5) for further details. | 26 May 2024 |
| Article 5(5)d | The requirement to justify that a patient group’s specific needs cannot be met or cannot be met at the appropriate level of performance by an equivalent device available on the market. | 26 May 2028 |
| Article 5(5) final paragraph | Member States’ rights to request information/restrict the manufacture and use of devices and inspect health institutions. | 26 May 2022 |
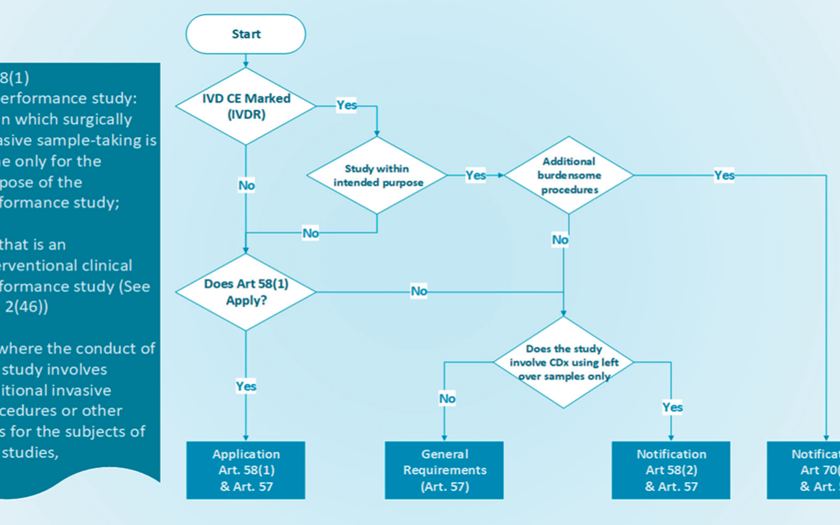
HPRA Decision Tree – IVDR Performance Study | 26 May 2022
A decision tree for IVDR performance studies was created to help sponsors determine whether IVDR regulations apply to their research, such as whether an application or notification is necessary. Refer to IVDR Chapter VI for more details.

IVDR Registration Requirements | 26 May 2022
The usage of the EUDAMED database to meet registration obligations is recognised by HPRA. Article 28 of the IVDR outlines the responsibilities of manufacturers, authorised representatives, and importers. To comply with the registration duties, Irish economic operators are encouraged to use the EUDAMED’s actor registration module. By completing the online registration form, distributors, health institutions, and production facilities can register with HPRA.
CZECH REPUBLIC
Information systems in the field of regulation of medical devices | 19 May 2022
The National Medical Device Information System (NISZP) published a document outlining the information systems utilised in the Czech Republic and the European Union to regulate medical devices. The document conveys the following information:
- the current medical device regulation in the Czech Republic,
- EUDAMED Database, its modules, and the deadlines
- UDI
- Agendas of the Medical Device Information System (ISZP)
The Czech version of the document can be found here.
European Commission implementing decisions on harmonised standards for medical devices published in the Official Journal of the European Union | 17 May 2022
The National Medical Device Information System (NISZP) issued the Czech versions of the EU’s decisions:
- 2022/729 of 11 May 2022 amending Implementing Decision (EU) 2021/1195 related to the standards for the quality management systems, and the application of risk management to medical devices
2022/757 of 11 May 2022 amending Implementing Decision (EU) 2021/1182 related to the standards for quality management systems, sterilisation, and risk management application to medical devices
FRANCE
European guideline on borderline products | 19 May 2022
Medical Device Coordination Group (MDCG) published guidance for the Borderline Products (Medical Devices and Medicinal Products). Borderline products are those for which it is unsure whether they fall under the MDR or the MPD from the beginning. EU Regulation 2017/745 is used for governing the medical devices in the EU. In contrast, Directive 2001/83/EC regulates the medicinal products for human use (MPD). This document clarifies these regulations with additional explanations and examples to promote the MDR’s consistent application. The publication provides definitions and examples and sections on substance-based devices, plant-based goods, and drug-medical device combinations.
Cosmetics – Production communication | 19 May 2022
Manufacturers of cosmetics shall communicate cosmetics production to the Ministry, either on their own or on behalf of third parties, using the new form made accessible, including extemporaneous manufacturing and small quantities.
Establishment of the national network for supervision | 02 May 2022
The decree for establishing the national network for supervision was published in the official gazette. According to current legislation, public and private health professionals must immediately report any significant accident to the Ministry of Health and the device’s manufacturer. The Ministry of Health then oversees the investigations conducted by the manufacturer or his authorised representative, analysing and monitoring the effectiveness of the corrective actions made. The network was set up to make it easier to share information on events and safety actions involving medical devices, in vitro diagnostic medical devices, and equipment included in Annex XIV of Regulation (EU) 2017/745. The surveillance system is maintained by continuously monitoring medical equipment incidences after being placed on the market. This information system gathers data on the following topics:
- local supervisor and regional manager contact information
- incident reports created by healthcare professionals
- extracts from incident reports provided by manufacturers
- security measures
RUSSIA
Roszdravnadzor gave recommendations on the operation and use of medical devices connected to the Internet | 19 May 2022
If a medical organisation lacks the resources to employ a secure data transmission network, the medical device must either operate as part of a separated technical network or data should be sent between various components of medical devices employing portable data storage devices. Furthermore, Roszdravnadzor stated that before upgrading the software on a medical device, it is required to ensure the development of backup copies of the software so that it may be restored to its initial state.
Resumption of supplies of imported X-ray film to the Russian Federation | 16 May 2022
The Federal Service for Surveillance in Healthcare held a working meeting with authorised representatives of foreign manufacturers of X-ray film. The daily monitoring of Roszdravnadzor in the country’s regions recorded the defector of this medical product due to the search for new logistics routes by film manufacturers.
The Ministry of Health will study the issue of lifting the ban on the export of medical equipment abroad | 16 May 2022
The Ministry of Health of Russia is working on the issue of lifting the ban on the export of medical equipment from the country for repair as per the First Deputy Minister of Health of Russia, Viktor Fisenko. He highlighted that it is sometimes necessary to export equipment or components for repair overseas. The lifting of such limitations is now being discussed with the Ministry of Economic Development and Roszdravnadzor.
Russia will create a digital service for online medical consultations | 16 May 2022
GLONASS JSC and Polymed will create a telemedicine platform GLONASS Zabota for online consultations with medical specialists, a wide range of laboratory tests, and the order of medicines. This platform will enhance the number of laboratory tests available to citizens of all Russian regions, including those in remote areas, both at stationary and mobile locations with home visits. The service’s users will be able to send the study results to any polyclinic in Russia. Secure data transmission channels of the era-GLONASS state information system will offer the legal meaning of the information, allowing test results to be verified and uploaded to the Unified State Information System in healthcare.
Import substitution in the market of dental materials | 16 May 2022
90% of supplies, tools, and equipment used in private dentistry come from foreign manufacturers. Still, many transactions have become unfeasible due to recent anti-Russian sanctions and logistical issues. Consumables such as dental fillings, braces, mouthguards, and other items have also increased in price. According to experts, Russia has its production. It may replace items from Europe and the United States by refocusing on the Southeast Asian, Chinese, and South Korean markets.
Increase in the production of domestic medical equipment| 12 May 2022
Manufacturers should be aware that, to expand the line and bring domestic products to market as quickly as possible, Roszdravnadzor must reduce the time it takes to issue registration certificates for medical devices, as well as the issuance of certificates of origin of goods and acts of expertise by the Chamber of Commerce and Industry of Russia (CCI). At the same time, medical equipment are already eligible for faster registration under a government directive.
UNITED KINGDOM
Notify the MHRA about a clinical investigation for a medical device | 19 May 2022
All deviations from the study must be reported to the MHRA as soon as the manufacturer becomes aware of them. Details concerning the nature of the deviation, when and where it happened, and any recommended corrective and preventative activities, should be included using the ‘live’ excel template for submitting.
Joint Statement on the launch of the negotiations for a Free Trade Agreement between the UK and Mexico | 20 May 2022
On 20 May 2022, Tatiana Clouthier, Secretary of Economy of the United Mexican States, and Anne-Marie Trevelyan, Secretary of State for the United Kingdom’s Department for International Trade, met in London. They announced the beginning of negotiation for a Free Trade Agreement between the United Mexican States and the United Kingdom in a joint statement. The first official round of the talks will occur in Mexico City in July 2022, followed by a second round in the autumn. This agreement will improve investment flows, strengthen trade in products and services, and encourage digital and cross-border trade.
SWITZERLAND
New regulations applicable to in vitro diagnostic medical devices as of 26 May 2022 | 26 May 2022
The Federal Council adopted the new In-vitro Diagnostic Medical Devices Ordinance (IvDO) and an amendment to the Ordinance on Clinical Trials (CTO-MedD) with Medical Devices on 04 May 2022. The new legal requirements take effect on 26 May 2022, coinciding with the EU’s implementation of the IVDR. Clinical trials with in vitro diagnostic medical devices will be governed by the Ordinance on Clinical Trials with Medical Devices (CTO-MedD) beginning on 26 May 2022, rather than the Ordinance on Clinical Trials (ClinO).
New rules for performance studies | 26 May 2022
Except for performance studies, the provisions of ClinO-MD apply to all performance studies as of 26 May 2022. To assess if a study project must be submitted to Swissmedic, a decision tree for categorising performance studies has been made accessible. Swissmedic and the competent cantonal ethics council must additionally approve performance studies based on this decision tree.
Status of the EU-Switzerland Mutual Recognition Agreement (MRA) for in vitro diagnostic medical devices | 25 May 2022
Switzerland has previously participated in the European Union’s internal market for in vitro diagnostic medical equipment under the medical devices chapter of the EU Switzerland Mutual Recognition Agreement (MRA). The medical devices chapter of the MRA provides the acceptance of conformity assessment certificates between the European Union and Switzerland based on the equivalence of Directive 98/79/EC and the applicable Swiss law. However, with the introduction of the new IVD Regulation (EU) 2017/746, the MRA has become obsolete from 26 May 2022, resulting in the following consequences:
- Swiss in vitro diagnostic medical device manufacturers will be classified as third country manufacturers in the European Union (EU)
- Conformity assessment bodies established in the EU must certify the conformity assessment procedure
- Certificates issued under the MRA by Swiss conformity assessment organisations will no longer be recognised as valid in the EU, even if granted before 26 May 2022
- Swiss manufacturers and third country manufacturers must designate an authorised representative established in the EU for placing the devices on the EU market after 26 May 2022
Medtech in Switzerland 2030 | 17 May 2022
On 17 May 2022, Swiss Medtech announced its vision for “Medtech in Switzerland 2030”, describing methods to maintain Switzerland’s appeal as a Medtech company destination in the future. Stable trade ties, open markets, innovation promotion, and reimbursement schemes tailored explicitly to the Medtech industry were discussed. The reform agenda is essential since the failure of institutional agreement discussions between Switzerland and the European Union (EU) has affected business competitiveness.
Approval of medical devices according to non-European regulatory systems | 06 May 2022
Switzerland only accepts medical devices for national supply that are CE marked and comply with the European Union’s (EU) medical device regulation. Additionally, Switzerland also accepts medical devices from non-European regulatory systems. This is especially true for medical devices that have been approved by the US Food and Drug Administration (FDA).
AUSTRALIA
Seasonal Influenza Rapid Antigen Self-tests and Combination tests | 27 May 2022
The document’s goal is to provide manufacturers and sponsors with guidance on the Therapeutic Goods Administration’s (TGA) expectations for clinical performance requirements (clinical sensitivity and specificity) and risk mitigation for in vitro diagnostic medical devices (IVDs) intended for use as self-tests for seasonal influenza and combination tests (for both COVID-19 and Influenza).
Personalised medical device framework | 20 May 2022
TGA uploaded a presentation on the medical device regulations’ overview.
Regulation of software-based medical devices | 03 May 2022
TGA provided provisional guidance to assist manufacturers and sponsors in understanding how the TGA interprets regulations and indicates how they can comply.
Unique Device Identification | 02 May 2022
TGA held a webinar for the Unique Device Identification on 19 April 2022. The presentation of the webinar #8 can be found here. TGA has been undertaking series of webinars on the UDI topics since 2021.
UNITED STATES OF AMERICA (USA)
Revocation of Authorisation of Emergency Use of an In Vitro Diagnostic Device for Detection and/or Diagnosis of COVID-19 | 24 May 2022
The FDA granted the Broad Institute an Emergency Use Authorization (EUA) for the CRSP SARS-CoV-2 Real-time Reverse Transcriptase (RT)-PCR Diagnostic Assay on 08 July 2020, subject to the provisions of the Authorisation. Later, on 04 April 2022, the institution requested revocation from the FDA.
Feasibility and early feasibility clinical studies for specific medical devices intended to therapeutically improve glycemic control in patients with type 2 diabetes mellitus | 06 May 2022
T2DM (Type 2 Diabetes Mellitus) is a metabolic condition characterised by high blood sugar levels, insulin resistance, and insulin deficiency. As a result, several medical device manufacturers and researchers are working to create therapeutic medical devices, including blood glucose monitors and insulin pens, pumps, and syringes, to help patients with T2DM improve their glycemic control. The FDA released a guidance document outlining its initial thoughts on feasibility and early feasibility clinical investigations for these medical devices. These medical devices are designed to lower glycated haemoglobin (HbA1c) levels in T2DM patients without using medications like insulin. Manufacturers are encouraged to submit a Pre-Submission to obtain detailed feedback on the clinical investigation of medical devices within the scope of this guidance that are intended to therapeutically improve glycemic control in patients with T2DM before initiating a pivotal clinical study or to receive additional feedback on the feasibility study design before starting a crucial clinical study.
Voluntary medical device manufacturing and product quality pilot program | 05 May 2022
In 2018, the FDA and the Medical Device Innovation Consortium (MDIC) conducted a voluntary pilot programme among medical device manufacturing sites. MDIC program activities and operations are moving into a permanent program, termed the Case for Quality Voluntary Improvement Program (CfQVIP), based on the pilot’s success. In preparation for the transition to a permanent programme, the FDA and MDIC cooperated to create a charter and a governing committee for CfQVIP. The FDA is currently developing a complementary policy for engaging with CfQVIP. When adopted, this FDA regulation will be based on the FDA’s experience with the Voluntary Medical Device Manufacturing and Product Quality Pilot Program.
Supplements for approved Premarket Approval (PMA) or Humanitarian Device Exemption (HDE) submissions during the coronavirus disease 2019 (Covid-19) public health emergency | 04 May 2022
The FDA issued a guidance document to give the policy to help address existing manufacturing limits or supply chain concerns due to the public health emergency created by COVID-19.
BAHRAIN
Assigning a Post marker surveillance officer | 12 May 2022
As part of the NHRA’s role in ensuring the safety of medical devices in the post-market phase, all healthcare facilities and authorised representatives are strongly encouraged to appoint a PMS officer who is responsible for medical device reporting in the post-market surveillance stage, including field safety notices, adverse events, and complaint reporting to the NHRA.
INDIA
Insertion of rule 43A in MDR 2017 for suspension and cancellation of license | 18 May 2022
The Ministry of Health and Family Welfare has released a Notification to the Official Gazette to amend the Medical Devices Rules 2017. Following consultation with the Drugs Technical Advisory Board and in the exercise of the powers provided by sections 12 and 33 of the Drugs and Cosmetics Act, 1940, the Central Government applies the following regulations to modify the Medical Devices Rules, 2017:
- These rules may be called the Medical Devices (Third Amendment) Rules, 2022
- They will take effect on the day they are published in the Official Gazette.
- Rule 43A – Suspension and cancellation of license for the entity failing to comply with the conditions of an Import License
PAKISTAN
Amendments in the Medical Devices Rules, 2017| 18 May 2022
On 27 April 2022, the Drug Regulatory Authority of Pakistan (DRAP) received approval from the Federal Government to amend the Medical Devices Rules, 2017. The document can be found here.





