The booming technological aspects have constantly challenged regulations established worldwide in the modern century because the regulations either inadequately govern novel technologies, or there always exists a scenario where the devices cannot be categorized within a given regulation.
Smart wearables are one of those aspects falling into such debate in recent times. Days are no longer from the fact that these smart wearables may draw strict regulations to be in place.
How are these general wellness devices regulated? What are these devices classified as?
Unlike medical devices, these devices are intended to be used for wellness reasons such as to improve the lifestyle by monitoring the user’s diet and exercise, enhancing physical activity etc., and they are ideally not intended to get involved in any diagnosis or therapy.
Intended Use
Any device is subject to a regulation based on its “intended use” declared by the manufacturer. Hence the manufacturer needs to pay utmost attention in declaring what their device supposed is to be used for real-life scenarios.
Smart wearables are often confused with medical devices, or sometimes they are by mistake considered medical devices if a vague intended use statement is written for the device.
Generally, smart wearables are low-powered electronic equipment intended to be worn by the user, such as wrist-worn, neckband, body-worn, adhesive to the skin surface to collect some data and transfer the information to a mobile device application.
Sensors and electronic components present in your smart wearables monitor your activities which could be determining the body’s orientation, climbing stairs, speed of walking or running, heartbeat, oxygen levels, respiratory rate, sleep patterns, etc.
Now the question here is, should one consider this a medical device when the device monitors HR, SpO2, and respiratory rates, which a physician usually monitors to determine the state of health and use these data to diagnose any physiological condition.
The answer is not straightforward, but there are smart wearables in the market that obtain FDA clearance that claims to perform certain medical purposes as the intended purpose. Such devices fall under the medical device category.
Hence, if the manufacturer claims that the device has a “medical purpose” in its intended use, the device falls under a medical device. Claims are critical in these cases.
How is my smart wearables regulated?
In Europe, there is no separate regulation released to date for smart wearables; however, depending on the nature of your device, it is mandatory to adhere to the applicable EU directive or EU Regulation. CE marking is mandatory for all categories of equipment entering the EU market.
It is essential to identify the applicable regulations for the device based on the nature and functionality of the equipment. For any electronic radio equipment, the following EU directives apply
· Radio Equipment Directive 2014/53/EU
· EMC Directive 2014/30/EU
· Low Voltage Directive 2014/35/EU
· RoHS and REACH Directive 2011/65/EU
· WEEE Directive 2002/92/EC
The below tables give a quick brief of key requirements applicable across the directives and this list is non-exhaustive:
Sections
Radio Equipment Directive
EMC Directive
Low Voltage Directive
RoHS and REACH
Essential Requirements
Article 3
Article 6 Annex I
Safety Objectives -Annex I
Article 4, Annex II Prevention
Technical Documentation
Article 21 Annex V
Annex II or III
Annex III
Article 6 Review of Restricted Substances
Conformity Assessment
Article 17, Annex II or III or IV
Article 14, Annex II or III
Annex III
Not applicable
Obligations for Economic Operators*
Applicable
Applicable
Applicable
Applicable
EU DoC
Article 18 Annex VI
Article 15 Annex IV
Article 15, Annex IV & Module A of Annex III
Article 13, Annex VI
Simplified DoC
Article 18 Annex VII
Not applicable
Not applicable
Not applicable
Field Corrective measures
Applicable
Applicable
Applicable
Applicable
*-Economic Operators consists of Manufactures, Authorised Representatives, Distributors and Importers
The above listed are very few directives applicable for a low-powered electronic radio equipment category of the smart wearable device. It is the manufacturer’s responsibility to identify relevant regulations suitable for the device and adopt the same to meet product compliance.
The directives and standards differ based on the type of devices, such as IT and telecommunication, digital, and audio/video radio equipment.
How to use Harmonized Standards to demonstrate compliance?
Harmonized standards are voluntary standards adopted by the EU commission and deemed acceptable to demonstrate product compliance. It is presumed that by adopting harmonized standards published by the EU commission for those directives, the manufacturer can claim product compliance to the respective directive.
The standards adopted for design and testing should comprise safety and functional performance relevant to the devices. It also must meet the essential requirements of the directives/regulations.
Accredited Laboratory Testing
Testing is an integral part of demonstrating product compliance. It is always advisable to approach an accredited third-party laboratory to perform the applicable tests for your product. Manufacturers’ internal test reports are rarely considered for compliance or sometimes a risky element to showcase such reports.
Significance of Technical Documentation
As stated above, RED, EMC and LVD directives mandate the manufacturer to maintain technical documentation of the product with a retention period of 10 years from placing the device on the market.
Instructions for Use (IFU) and Labelling
Manufacturers are obliged to supply adequate information on the device’s safe usage and the device. The accompanied instruction and safety information must be in a language that can be easily understood by the user and be in the official languages of the member states. The safety information of device interference characteristics must be disclosed for radio equipment that intentionally emits radio waves.
Declaration of Conformity
The manufacturer must provide the DoC with each device sold in the market, and it needs to be in the official language of the member states. In cases where more than one directive or regulation is applied to the device, the contents of the DoC must include all the elements listed in each of those directives. In certain directives, a simplified DoC is also required to be mentioned in the IFU, where it states the location of the updated DoC on the manufacturer’s website.
Conclusion
Regulations in recent years are becoming increasingly stringent towards safety. As these devices bring in more advancements and are closely engaged with the human lifestyle to promote health and wellness, there must be careful consideration in bordering these products from being falling into the medical devices category. Eventually, be it a medical device or any equipment, it is the ultimate responsibility of the manufacturer to produce safe products.
FAQs
Does the EU maintain a list of harmonized standards?
How does the EU regulate Data protection and privacy of the data?
General Data Protection Regulation applies to devices that collect and process the users’ personal data. European Union has sought to ensure the protection of this right through legislation. The vital regulatory principles of this GDPR are data protection principles, accountability, Data security, Data protection by design, consent, and people’s privacy rights.
Does US FDA have any regulation on smart wearable technologies?
FDA does not regulate general wellness devices, but it has released general guidance on the “general wellness: policy for low-risk devices” to provide clarity to industry and FDA staff on the Center for Devices and Radiological Health’s (CDRH’s) compliance policy for low-risk products that promote a healthy lifestyle.
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.
This guidance is applicable to manufacturers placed within India. Get detailed information about CDSCO registration process.
Regulation Medical Devices Rules, 2017
Important Forms
License to manufacturer Notified Medical Devices
License in Form MD-5 or 6 for Class A or Class B
Application form Form MD-3 or 4 for Class A or Class B Rule 20
Rule 20(4) and 20(6) for Class A or Class B
State Drug Licensing Authority
License to manufacturer Notified Medical Devices
License in Form MD-9 or 10 for Class C or Class D
Application form Form MD-7 or 8 for Class C or Class D Rule 21
Rule 25(1) for Class C or Class D
Central Licensing Authority
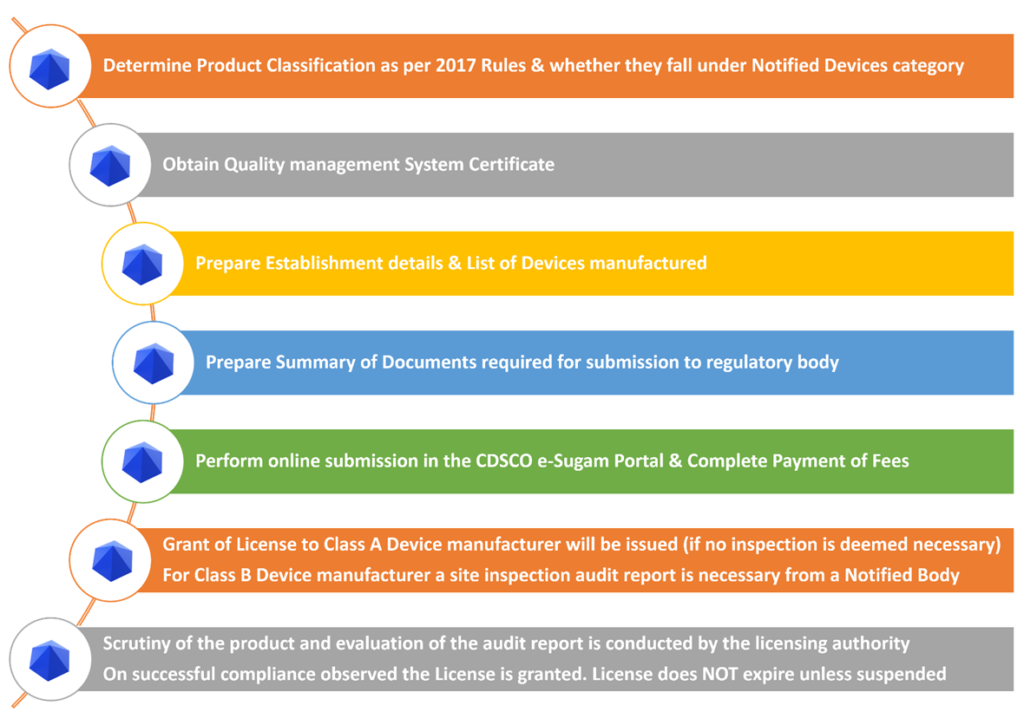
Registration Process
Product Classification
Determine Product Classification as per India Medical Devices Rules, 2017 if they fall under the below as per Part I of first Schedule
Class A, B, C, D
Determine if the classified device falls under Notified Medical Devices or Non-Notified Medical Devices.
Quality Management System Certification
Establish and obtain a QMS certificate from a Notified Body (QMS defined in Fifth Schedule).
Perform Medical Device compliance testing as per BIS Standards. If no relevant BIS standard(s) are available, the manufacturer must choose to follow ISO/IEC standard compliance relevant to the medical devices
e-SUGAM Online Submission Requirements
The next significant step is to prepare the documents for the online submission as per Fourth Schedule Part I, II & III. The applicable documents are dependent on the class of device in the application.
Name and address of the company along with the address details of the site of manufacturing
Details of medical device:
Generic Name
Model No.
Intended Use
Class of medical device
Material of construction
Dimension
Shelf life
Sterile or Non-Sterile
Brand name if registered under the Trademarks act, 1999
ISO 13485 Certificate
Free Sale certificate from the country of Origin (not applicable for those manufactured within India)
Undertaking duly signed by the manufacturer stating that the information furnished by the applicant is true and authentic
Other required documents are listed at the end of this article
Online Registration Process
Steps to Start CDSCO License Application Process
It is required to appoint a competent technical staff who meets the competency listed in Rule 22(ii).
Go to CDSCO home site >> Online System for medical devices
Click on Login/Register to arrive at the Registration homepage
Select from the Purpose >> For Manufacture of Medical Devices
Fill in the complete online application form
Upload all the documents as per in the checklist
Make the payment in the same portal (Link to Fee details are given at the end of this article)
Once the application is filled and complete, Click Submit
A confirmation email will be received to the registered email address
Track the application in the online portal
Timelines for Granting CDSCO Manufacturing License
For Class A
Within 45 days from the date of receipt of application the State Licensing Authority grants the license in Form MD-5 or Form MD-6 or rejects the application
For Class B
Within 90 days from the date of receipt of the application, the Notified body shall conduct an inspection of the manufacturing site
Within 30 days, the Notified body shall submit the audit report to the licensing authority
Within 20 days, the SLA scrutinises the documents, audit report and grant License in Form MD-5 or MD-6
For Class C or D
Within 45 days from the date of receipt of application, the CLA shall perform scrutiny of the application and technical documents using an expert
Within 60 days, a site inspection is carried out by the licensing authority comprising of experts (as mentioned in Rule 23) & the inspection report is sent copy to the applicant.
Within 45 days, the CLA scrutinises the documents, audit report and grant License in Form MD-9 or MD-10
Other Documents required for submission
Power of Attorney (provided in MDR -Fourth Schedule, Part 1) To be authenticated in India either by a Magistrate of First Class or by the Indian Embassy in the country of origin or by an equivalent authority through apostille.
Device description intended use of the device, specification including variants and accessories.
Material of construction.
Working principle and use of a novel technology (if any).
Labels, package inserts (IFU, etc.,), user manual, wherever applicable
Summary of any reported Serious Adverse Event in India or in any of the countries
Site or plant master file as specified in Part III Appendix I of MDR
Device Master File Part III Appendix II (Risk assessment
Constitution details of the firm (of the domestic manufacturer or authorised agent)
Essential principles checklist
Undertaking signed by the manufacturer stating that the manufacturing site is in compliance with the provisions of the Fifth Schedule
Copy of latest inspection or audit report carried out by Notified bodies or National Regulatory Authority or Competent Authority within last 3 years, if any.
Navigate in Menu options such as Alerts, News, Public Notices and Gazette Notifications to find all the recent updates and releases
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.
A Label is the written, printed, or graphic information that goes on the packaging of the medical device.
Instructions For Use (IFUs) or Package Insert is the essential information accompanying the medical device for its safe and effective use by the user. It can be a single to multiple-page document.
Labelling is the content that goes on the Label or IFUs.
What are the minimum requirements for labeling?
The ISO has published many standards applicable to the medical device industry. Some of them are as below:
Standard Number
Standard Name
ISO 18113
In vitro diagnostic medical devices – Information supplied by the manufacturer (labelling) – Part 1, 2, 3, 4 and 5
ISO 28219
Packaging – Labelling and direct product marking with linear bar code and two-dimensional symbols
Medical devices – Information to be supplied by the manufacturer
ISO 14025
Environmental labels and declarations – Type III environmental declarations – Principles and procedures
ISO 14021
Environmental labels and declarations – Self-declared environmental claims (Type II environmental labelling)
ISO 14020
Environmental labels and declarations – General principles
ISO 22742
Packaging – Linear barcode and two-dimensional symbols for product packaging
There are more specific product-oriented labelling standards available.
ISO 20417 has defines information to be disclosed by the manufacturer. Every medical device manufacturer, distributor, importer, or Authorized Representative is bound to comply with the standard before placing the device on market. The requirements are as follows:
Information on Label
Manufacturer details – Trade Name, address, country
Product description.
Product identification – model or catalogue number, Lot number, serial number, expiry date, UDI,
Storage instructions
Operating instructions
Warning or precautions
Presence of any harmful substances (>0.1% w/w), biological origin substances, medicinal substances, nanotechnology materials
Electronic IFUs (if available)
Mention of: Single-use/ Single patient multiple-use / Reuse / Limitation on reuse
If Sterile and method of sterilization
Explanation of safety-related colours
Information on Packaging
Name and address of the manufacturer or an authorized representative
UDI
Production controls – lot number, serial number, expiry date
Model number, catalog number, commercial name
Mention of: Single-use/ Single patient multiple-use / Reuse / Limitation on reuse
Storage or special handling requirements
Any special requirements for battery-powered medical device
Contraindications, warnings, or precautions
Information in IFUs
General information (as above)
Intended Use of the medical device
Safety information
Performance of the medical device
Any residual risk associated with the use of the medical device or its accessory
Any known contraindications
Document control number of the IFU
Safe disposal information
Any specific instructions for handling or preparatory treatment
Any warnings, precautions, or limitations
If any accessories or indicators are provided along with the device, instructions on their use to be provided in the IFU.
Technical description
The harmonized ISO standard makes sure true and uniform information is conveyed to a lay/common person.
Global Labelling Requirements
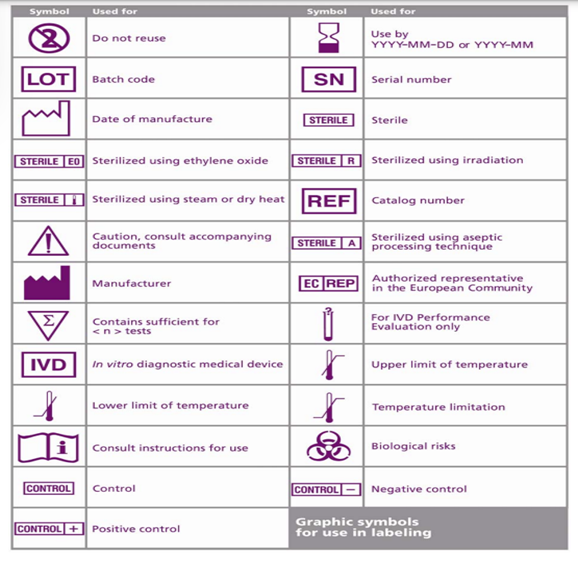
Most countries have a mandatory requirement for the IFUs or Labels in their local language. To streamline this requirement, ISO 15223 standard provides a list of signs and symbols that depict common terms such as Manufacturer, Lot number, storage conditions, Expiry, eIFU and many more.
The uniform symbols help in identifying the necessary information without the language barrier. Another advantage is it saves significant label space.
FAQs
Is it necessary to follow the ISO standards?
It is advisable to develop a medical device in compliance with the applicable harmonized standards. This shall favor in smooth marketing of the product along with its competitors.
Is it necessary to brief the symbols in IFU when symbols from standards are used?
Yes, it is required to brief every symbol in the IFU that is used on the label of the product.
Can a distributor or an importer label be affixed separately apart from the main label?
Yes, it is also allowed to affix these labels separately on the product. This is because one manufacturer may have several distributors or importers within EEA.
Is it necessary to create dedicated labels for accessories of medical devices?
Yes, it is. Not every time the accessory is shipped along with the medical device and it is required to identify them with appropriate labels.
If the manufacturer wants to provide an eIFU how to indicate this on the label?
Firstly, not all the medical devices are eligible for eIFU provision. Regulation 207/2012 states what are the categories of MDs that are eligible for eIFU.
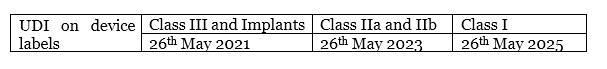
What is the deadline to implement UDI carrier on device labelling?
Article 123.3.f states these timelines as:
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.
ISO – International Organization for Standardization, is the international, non-governmental body for drafting and establishing technical and non-technical standards.
These standards are developed by different committees within the International Organization for Standardization. Having around 165 member states, with one representative from each, International Organization for Standardization is a global entity catering to the needs of industry requirements.
Are ISO standards important?
The International Organization for Standardization medical device standards are the Bible for many countries, especially ones which do not have predefined regulations or processes.
In addition to general standards, ISO also publishes product-specific guidance such as for Implants, Orthopedic, Medical Electric Equipment, and many more.
Global International Organization for Standardization Requirements
In Europe, the European Commission has the Medical Device Regulation MDR 2017/745 and In-vitro Diagnostic Device Regulation IVDR 2017/746.
These regulations provide a detailed framework for introducing a medical device in the European market. However, in addition to that, certain International Organization for Standardization may also be referred to for ensuring a better-quality product.
Some of the many popularly used standards include:
ISO 14971:2019 Medical Devices – Application of Risk Management to medical devices
ISO 15223-1:2021 Medical devices – Symbols to be used with information to be supplied by the manufacturer – Part 1: General requirements
IEC 60601-2-83 Medical electrical equipment – Part 2-83: Particular requirements for the basic safety and essential performance of home light therapy equipment
IEC 60601-1 Medical electrical equipment – Part 1: General requirements for basic safety and essential performance
The European Commission also has Harmonized Standards, developed by European Standards Organization CEN, CENELEC, or ETSI, per the international standards.
It provides a list of the applicable harmonized standards for enhanced product safety and quality.
In the USA, the US Food and Drug Administration (FDA) has a Code of Federal Regulations (CFR) and Guidance.
CFRs are legally binding. Manufacturers must comply with the requirements of CFR
The guidance provides Agency’s thinking on regulatory issues. They are NOT legally binding
In addition to these, the FDA also accepts certain recognized consensus standards from different organizations such as International Organization for Standardization, CLSI, ANSI, IEC, CEN, etc.
These standards may be used to justify a Declaration of Conformity for a product. The widely accepted medical device International Organization for Standardization are, but are not limited to:
ISO 10993 – Biological Evaluation for Medical Devices
ISO 14160 – Sterilization of Healthcare Products
ISO 11737 – Sterilization of Medical Devices
In Canada, the Standards Council of Canada (SCC) is the International Organization for Standardization member body. Similar to the US FDA, the Therapeutic Products Directorate (TPD) of Health Canada periodically releases a list of acceptable international or national standards for medical devices.
Manufacturers can use these recognized standards in conjunction with the Health Canada’s Medical Devices Regulations (SOR-98/282) and the Guidance Documents, to prove product conformity and safe use in the market.
China‘s National Medical Products Administration (NMPA) is developing indigenous standards that more closely align with those of ISO. Biocompatibility testing is one avenue where the scope and requirements for China are more than that of the US/EU.
Hence, NMPA has developed various biocompatibility testing standards which are to be used in addition to the International Organization for Standardization standard.
For the rest of the world’s medical device industry,
India encourages International Organization for Standardization certification for all its industries. The medical sector must be International Organization for Standardization 13485 compliant while the pharmaceutical sector must be ISO 9001 compliant for Quality Management Systems, in addition to other relevant and applicable International Organization for Standardization.
Japan’s The Japanese Industrial Standards Committee (JISC) is an International Organization for Standardization member body. The regulatory authority, Pharmaceutical and Medical Device Agency (PMDA) revised its Ordinance No. 169 in 2021 to closely align with the International Organization for Standardization 13485:2016 standard. The transition period is 3 years and must comply by March 25, 2024
For the Korean regulatory authority, aligning the requirements of Korean Good Manufacturing Practice (GMP) to that of International Organization for Standardization 13485:2016 is believed to be a step closer to entering the Medical Device Single Audit Program (MDSAP)
Russia’s Federal Service for Surveillance in Healthcare (Roszdravnadzor) is known to accept International Organization for Standardization 13485:2016 certification. Information on acceptance of other International Organization for Standardization cannot be confirmed. It does not accept market approvals in the US, EU, or other countries as a reference for market authorization in Russia
Australia’s Standard Australia is a member of the International Organization for Standardization, IEC, and ICSID. It strongly encourages the use of international standards, except where their use is ineffective or inappropriate and does not develop any national Australian standard for which there is already an international standard in existence. In 2019, TGA published Therapeutic Goods (Conformity Assessment Standard for Quality Management Systems) Order 2019which provides a list of applicable conformity assessment standards.
Brazil’s ANVISA accepts Good Manufacturing Practices (GMP) along with the International Organization for Standardization 13485
FAQs
Can QMS be established solely based on ISO standards?
For countries that do not have their own QMS regulations, the ISO standard can be used as a reference. For countries with established local regulations, and that accepts ISO, both ISO standard and local/national regulations must be considered.
Are ISO standards freely available?
No. ISO standards are available for purchase from the ISO official website. However, they do have FREE read-only formats available.
Comparing ISO standards to local regulations, which one takes precedence?
The local or national regulation always takes precedence over the ISO standard.
Can the manufacturer use an older version of an ISO standard for compliance?
No. Manufacturers must make sure they comply with the active or most recent version of the ISO standard. This is not restricted just to ISO standards but applies to National regulations too. Manufacturers must keep their QMS up to date with the latest requirements of the industry. The ideal way to be updated is to refer to the latest version of any Standard or Regulation.
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.
Quality Management Systems (QMS) are defined by the MDR as formalised systems that document processes, responsibilities, and procedures to ensure and continually improve the standard of company activities.
To obtain CE marking and market devices in European Union, an effective Quality Management System that complies with EU MDR 2017/745 is must.
Article 10 of EU MDR 2017/745 explains the general obligation of manufacturers to place their products in the EU market. Article 10 of the EU MDR 2017/745 also comprises the requirements for implementing and maintaining a Quality Management System capable of addressing particular issues to ensure device quality and safety.
Manufacturers of devices are expected to establish, document, implement, maintain, and keep up to date all the device documents to continually aid the improvement of the quality management system.
QMS for EU MDR
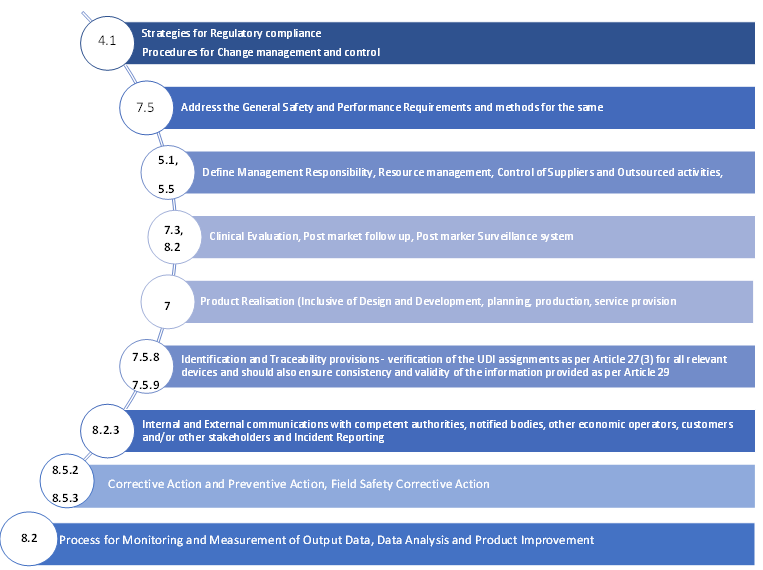
Quality Management System (QMS) must address at least the following:
A strategy for regulatory compliance, which includes conformity assessment procedures and procedures for change management and control for the device
The system must consist of general safety and performance requirements and methods to address the same
The system must define the management’s responsibility
The system must include resource management methodologies which should contain information on the selection and control of suppliers and sub-contractors
The system must include risk management methodologies as per Section 3 of Annex I
The system must include clinical evaluation data along with post-market clinical follow-up (PMCF) as per Article 61 and Annex XIV
The system must include data on product realization, which should involve information such as planning, design, development, production, and service provision
The system must ensure the verification of the UDI assignments as per Article 27(3) for all relevant devices and should also ensure consistency and validity of the information provided as per Article 29
The system must contain data on the setup, implementation, and maintenance of a post-market surveillance system as per Article 83
The system must include communication protocols with competent authorities, notified bodies, other economic operators, customers and/or other stakeholders
The system must include methodologies for reporting serious events and field safety corrective actions in the context of vigilance
The system must also include methodologies of management of corrective actions and verification of their effectiveness
The system must include data on processes for monitoring and measurement of output data, data analysis and product improvement
Mapping of ISO 13485:2016 clauses to the MDR QMS requirements
FAQs:
How will a QMS be assessed under the EU MDR?
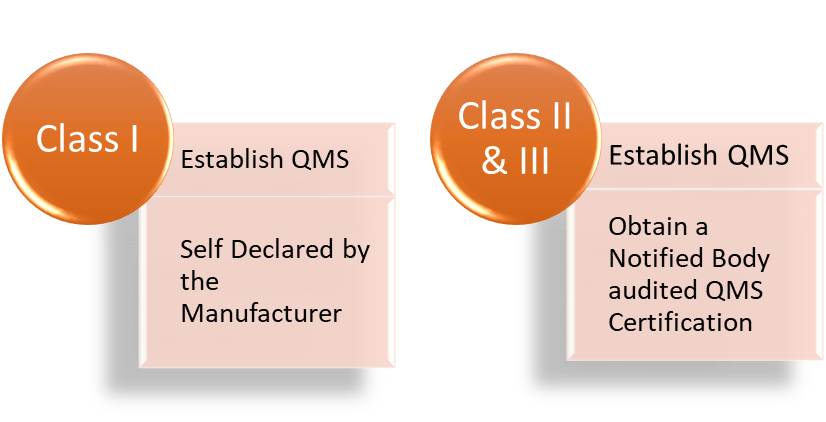
The quality management system of a manufacturer will be evaluated as part of the EU MDR Annex IX to XI- conformity assessment procedures. A Notified Body will do conformity evaluation for all devices except Class I.
Can ISO 13485:2016 QMS certificate holders bypass or claim EU MDR compliant QMS by themselves?
No, to be compliant with EU MDR the manufacturer is required to obtain a QMS assessment w.r.t Article 10(9) by a notified body in case of Class II & III. For a class I, the manufacturer must establish a corresponding QMS and include the same declaration in the self-declared DoC
Is 13485:2016 mandatory for MDR in addition to Article 10(9)?
No, the ISO 13485:2016 is not mandatory. However, the Quality Management System ISO 13485:2016 ensures entry to various global markets.
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.
EU MDR – Article 117 – The French Poly Implant Prostheses (PIP) breast implant controversy prompted medical rules to be revised and formed new Medical Devices and In-Vitro Devices Regulations to safeguard public health.
As per the European Medicines Agency (EMA) definition, a medicinal device is a substance or combination of substances intended to treat, prevent or diagnose a disease, or restore, correct or modify physiological functions by exerting a pharmacological immunological or metabolic action.
Medical devices, or device parts, are increasingly being produced for use with medicinal items, which might range from a basic prefilled syringe to more complex autoinjectors to medical devices incorporated as sensors in tablets.
Regulatory bodies have created specialised capabilities and requirements over the last decade due to the increased integration of medicines and devices seen in the latest generation of combination products.
To achieve fast and proper market access for new combination products, manufacturers must adequately understand the unique requirements in each country, and the development of combination products entails a specific pattern of engagement between manufacturers and regulatory bodies.
Based on their primary mode of action, products that combine a medicinal product (or substance) and a medical device are governed by Regulation (EU) 2017/745 or Directive 2001/83/EC.
How is a product categorised as a medicinal product or a medical device based on its mode of action?
As per the EU MDR 2017/745, the product is controlled as a:
medical device when the action of the medicinal substance is auxiliary to the action of the medical device,
medicinal product when the action of the medicinal substance is prominent and not supplementary to the action of the medical device.
For example, an insulin injector pen’s principal action is insulin administration. As a result, the injector pen will be classified as a medicinal product.
The necessity of a declaration of conformity and an EU certificate issued by a notified authority for the evaluation of compliance of the device part and the Drug-Device Combination marketing authorisation dossier is the most significant change brought about by Article 117 of EU MDR.
Suppose these conformity assessment documents are excluded from the dossier. In that case, the applicant will be asked to give an opinion from a notified body on the device part’s compliance with the relevant requirements of EU MDR Annex I.
A notified body is a conformity assessment service provider appointed by the National Competent Authority to assess the conformity of medical devices before being placed on the Union market.
When the device is integrated with a medicinal substance where the action of that substance is principal, or when the medical device administers a medicinal product forming a single integral product, such devices are considered an integral product (combination of medical device and medicinal product).
They shall be governed by Directive 2001/83/EC or Regulation (EC) No 726/2004 and the relevant general safety and performance requirements set out in Annex I of Regulation (EU) 2017/745.
The devices must meet the following criteria before they may be placed on the market:
The device and the medicinal product should be combined into a single integral product
The single integral product should not be reused
The single integrated product is only meant to be used in combination
Marketing Authorisation Applications (MAA)
An application by the Marketing Authorisation Holder (MAH)/applicant to a European regulatory body seeking authorisation to commercialise a pharmaceutical inside the European Union is known as a Marketing Authorisation Application (MAA).
Once obtained, the centralised marketing authorisation is valid in all EU Member States and EEA-EFTA countries (Iceland, Norway, and Liechtenstein).
According to Annex I of Regulation (EU) 2017/745, the MAA shall provide the following for a combination device starting on 26 May 2021:
Declaration of Conformity, and the CE certificate issued by a notified body for the device part having a CE mark
An opinion from a notified body on the conformity of the device part, if it does not hold a CE mark, Declaration of Conformity, and the CE certificate irrespective of the class of the device
Additional information of the benefit/risk assessment of the medicinal product when requested
Where a CE mark has not been issued to the device component on its own, the manufacturer must reach out to a NB and obtain their opinion on the device conformity and provide the NBO report in the MAA
In case the documents mentioned above are issued following the Medical Device Directives 93/42/EEC or 90/385/EEC, then they are still valid and can be used to support the requirements throughout the transition period, which lasts until 26 May 2024.
According to the European Medicines Agency (EMA) or the National Competent Authority (NCA), these documents must be provided in the initial MAA for the medicinal product, and if not, they must be produced before an opinion on the medicinal product application may be granted.
Failure to submit the appropriate documentation may cause the evaluation period to be delayed.
If any changes to the design, intended purpose of the device part, or a new version are launched after the Marketing Authorization has been granted to the manufacturer, the necessary declaration of conformity, CE certificate, and notified body opinion should be submitted to the EMA/NCA.
If the revisions influence the device’s quality, safety, or efficacy, the applicant/Marketing Authorisation Holders (MAH) must file a variation application, consulting the Medicines Competent Authority that gave the marketing authorisation (if the change is unclear).
As part of regulatory compliance, the relevant member states require the Mutual Recognition Procedure (MRP)/Repeat Use Procedure (RUP) application after submitting the variation application.
The dossiers for this application include the General Safety and Performance Requirements (GSPR) of the MDR and Annex I of Directive 2001/83/EC, Section 3.2’s point 12 of Directive 2001/83/EC as amended by Article 117 of the MDR, and supporting documents like the declaration of conformity, certificate of conformity or notified body opinion.
The European Public Assessment Report (EPAR) contains information on the submission and evaluation of materials, procedures, findings, and conclusions of the marketing authorisation application (MAA) to the EMA/NCA.
Co-Packaged Medical Devices (EU MDR – Article 117)
If a medical device is provided as part of the secondary packaging of a marketed medicinal product (co-packaged) and does not form an integral part of the medicinal product, the Marketing Authorisation Holder/applicant must ensure that their co-packaged medical device is CE marked, meets the MDR’s general safety and performance requirements, and is entirely compliant with the MDR before it can be placed on the market. Spoons, measuring cups, inhalers, and spacers are co-packaged medical devices. MDR 2017/745 must be followed by self-CE marked Class I devices.
Article 117 of the Regulation requires Notified Body involvement for European market authorisation of a medicinal product that incorporates an integral medical device or drug-device combination product. The EMA has released an FAQ to help the combination products’ manufacturers understand Article 117 requirements better. Manufacturers must keep in mind that the summary report of NBO will be issued by the notified body only after they verify if all the GSPR requirements are met adequately. Based on their opinion report, the medicines agency decides on the products’ MAA.
FAQs
Is the NBO report mandatory to be submitted to the MAA?
There are 2 ways to do this: 1. If the device is CE marked already, the manufacturer must submit a DoC and a copy of the CE certificate for the device. 2. If the device is not CE marked, obtain the NBO report (assessment of GSPR) and submit it to the authority along with your MAA application
What is a Single Integrated Drug-Device product?
If a medical device used to administer a medicinal product is placed on the market in such a way that the device and medicinal product form a single integral product which is intended exclusively for use in the given combination and which is not reusable, that single integral product shall be governed by Directive 2001/83/EC or Regulation (EC) No 726/2004, as applicable.
What are a few examples of Drug-Device Combination products?
Pre-filled syringes, inhalers, pre-filled pens, an antibody combined with a therapeutic drug, transdermal patches, and insulin pump. Co-packaging examples are a Drug or vaccine vial packed with a syringe and a dose-dispenser medication.
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.