Switzerland is a premier destination for medical device manufacturers relying on sophisticated micromanufacturing or advanced precision techniques. This appeal is evident in Switzerland’s choice of EMEA headquarters for industry leaders.
The country boasts one of the world’s highest proportions of GDP derived from the manufacturing sector, driven by traditional watchmaking, MEMS, and Medtech industries. These sectors have cultivated highly industrialized, digitized precision clusters ideal for crafting precise medical and orthotic devices.
The primary drivers compelling medical device manufacturers to opt for Switzerland include a technically adept workforce groomed by the country’s dual education system, ensuring proficiency in operating complex machinery.
Moreover, the attraction lies in Switzerland’s pragmatic, industry-tailored regulatory environment and an appealing tax system catering to foreign companies.
Medical Device Manufacturers – Pioneering Regulations, Efficiency, and Innovation
Many view Switzerland’s vocational training and close collaboration with research institutions and universities as facilitating quicker product validation and market entry.
While cost is often a reason for considering manufacturing or R&D elsewhere, numerous U.S. executives in Switzerland argue that the advantages surpass any increased expenses. Their skilled labour force proficiently handles advanced machinery, leading to fewer errors and ultimately lower costs.
Being near public authorities is advantageous, as these specialized hubs are usually geographically reachable. However, the ease of interaction with these authorities and an appealing tax system benefiting Switzerland as a European base further contributes to its attractiveness.
Testing, Scaling, and Collaboration
Switzerland offers an ideal setting for testing and expanding your business. Whether you’re experimenting with a new device or concept or engaged in the R&D phase, companies can swiftly progress from concept validation to commercialization due to an efficient bureaucracy and a tendency to avoid the usual bureaucratic obstacles in other nations.
Collaborating with Swiss corporations, startups, universities, and research institutions aids companies in extending and adjusting their businesses beyond U.S. borders, benefiting both sides.
Moreover, scaling becomes more manageable—obtaining permits and planning for new facilities is expedited and simplified.
Medical device nomenclatures are those products used to prevent, diagnose, treat, and monitor the many diseases known to humankind. Medical devices and medicines play an equally important role in treating human beings.
To learn more about medical devices, read our article on the definition of a medical device. This article discusses the nomenclature of medical devices and examples of these.
What is the Nomenclature of Medical Devices?
To simply put it, the nomenclature is the naming of a medical device. Although medical devices are classified into different risk classes, they should be named so that it is universally identified. Standardised nomenclature facilitates this easy identification.
A medical device nomenclatures is needed to simplify trade and tracking among the different regulatory authorities, Ministries of Health, and other organisations that regulate medical devices.
A standardised medical device nomenclatures aids in the following aspects:
Grouping and classification of medical devices.
Registration under different regulatory bodies or Ministries of Health
Streamlined procurement and distribution
Grouping of medical devices in various electronic health records and medical device databases
Vigilance reporting, field safety and post-market surveillance
The different medical device nomenclatures available are as follows:
GMDN or Global Medical Device Nomenclatures.
EMDN or European Medical Device Nomenclatures
UMDNS or Universal Medical Device Nomenclatures System
Other nationally developed nomenclature systems
Global Medical Device Nomenclatures (GMDN)
About 10% of countries use Global Medical Device Nomenclatures worldwide. It is a system of internationally accepted descriptors used to identify medical devices. The GMDN Agency manages GMDN codes, a non-profit organisation.
GMDN is a 5-digit code containing the following information:
GMDN Term Name: Anaesthesia ventilator
GMDN Code: 34851
GMDN Definition: A mains electricity (AC-powered) stand-alone, automatic cycling device used to assist and control alveolar ventilation during general anaesthesia and is compatible with inhaled anaesthetic agents. It has fewer functions and is less complex to operate than an intensive care ventilator but adequately meets the patient’s ventilation needs for oxygen (O2) and carbon dioxide (CO2) exchange to maintain normal blood gas concentrations. The device provides a mechanical means to deliver the breathing gas to the patient in a controlled pattern. It is equipped with alarms to warn of changes in respiration or the onset of unsafe operating conditions.
GMDN was introduced for a variety of regulatory purposes. GMDN is based on the ISO 15225: Medical device nomenclature data structure’. Read more about the frequently asked questions about GMDN here.
European Medical Device Nomenclature (EMDN)
European Medical Device Nomenclature or EMDN is introduced due to Article 26 of EU Regulation 2017/745 of Medical devices and Article 23 of EU Regulation 2017/746 of in-vitro diagnostic medical devices.
Like GMDN, it plays a considerable role in device nomenclature and serves various regulatory purposes. One of the primary uses is while registering a medical device in EUDAMEDwhere it is closely linked to UDI-DI.
Structure of EMDN
The European Medical Device Nomenclature is characterised by its alphanumeric structure and is established in a seven-level hierarchical tree where it clusters medical devices into three primary levels:
Categories: the first hierarchical level – alphanumeric.
Groups: the second hierarchical level – 2 numbers indicating group.
Types: the third hierarchical level – a series of numbers 1,2,3,4 and 5.
EMDN was adopted from the Classificazione Nazionale Dispositivi medici (CND) classification. The EMDN can be accessed at the EMDN list. European Medical Device Nomenclature categorises into three primary levels, categories, groups, and types.
A category comprises several groups composed of various kinds of medical devices.
Source: https://webgate.ec.europa.eu/dyna2/emdn/. In the above image of EMDN, ‘A’ is the category, ‘A01’ is Group and ‘A0101’ to ‘A0199’ are the types of medical devices.
Universal Medical Device Nomenclature System (UMDNS)
Universal Medical Device Nomenclature System or UMDNS was developed by the Emergency Care Research Institute (ECRI). Many nations have adopted this standard around the world. UMDNS is used in inventory control, work order control, and regulatory systems applications.
It is a 5-digit code unique code and a term for different types of medical devices. One can find the UMDNS code list here. UMDNS is updated monthly.
CND Nomenclature
CND nomenclature or ‘Classificazione Nazionale Dispositivi medici’ was developed by Italian Ministry of Health. In addition to Italy, it is also used in Portugal and Greece.
The guidance document on CND nomenclature explains the basic principles and structure of CND, which also applies to EMDN as EMDN was adopted from CND nomenclature. Following this, medical devices are clustered into three levels:
Category
Group
Type
FAQs
Is UDI the same as GMDN?
Both Unique Device Identification System (UDI) and GMDN are used in device identification. However, the two have some fundamental differences. UDI is inferior because of its lack of unity. It does not have a structure; therefore, device identification becomes more difficult with UDIs. Nonetheless, it is an effective tool for the traceability of medical devices. FDA utilises UDIs for medical device identification. The user guide to GUDID explains how the UDIs are managed in US FDA’s database, GUDID.
Can EMDN be accessed free of charge?
The EMDN is accessible to all stakeholders- free of charge. Hence, it can be utilised by a non-exhaustive list of stakeholders such as manufacturers, patients, research organisations, practitioners, hospitals, etc. The EMDN can also be downloaded from here.
Is there a guidance document that helps economic operators to map the EMDN information into the forthcoming EUDAMED database?
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.
Written, printed, or graphic information appears either on the device itself or on the packaging of each unit or the packaging of multiple devices (Art. 2 point 13 MDR). Timeline for Information on Indication of CH-REP “on the device” or “in the accompanying document.”
The provision for indication of CH-REP comes into effect on 26 May 2021 and is timeline based, applicable to devices of all classes (Class I, IIa, IIb, III).
For imported devices, the CH-REP should be indicated according to the below table:
Device Class
CH-REP Symbol / Indication to be placed on
Class I MDR Device
With effect from 26 May 2021 Until 31-Jul-2023Where: Either on the label (or) On the Document accompanying the device After 31-Jul-2023Where: On the label
Class IIa, IIb, III MDR Devices
With effect from 26 May 2021 Where: On the label
MDD/AIMDD Devices with EU/EEA manufacturer or EC-REP
With effect from 26 May 2021 Where:MDD – On label (or) Instructions for Use (or) In Document accompanying the device AIMDD – On sales packaging (and) Instructions for Use (or) In Document accompanying the device
MDD/AIMDD Devices without EU/EEA manufacturer or EC-REP
With effect from 26 May 2021 Where:MDD – On label (or) Instructions for Use AIMDD – On sales packaging (and) Instructions for Use
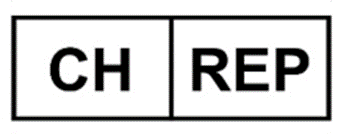
“CH-REP” Symbol
Representation / Usage of the “CH-REP” Symbol on the Device packaging
The name of the Swiss AR must be available on the product label.
The Name and Address of the Authorised Representative (AR) must appear adjacent to the symbol.
The mentioned Address must enable provision to contact the Swiss AR with sufficient contact details.
The symbol’s size and name are not defined; however, they must be adequately visible to the naked eye.
Symbols to Use
Where symbols are used, they must confirm to the harmonised standards. Swiss medic rule allows the sole use of the symbol without any description of the symbol explained in the product documentation enclosed.
Other CH-REP Representation(s)
Instead of the symbol, Swiss medic also permits the indication as stated below:
“CH-Authorised Representative”
“CH-REP”
“Authorised Representative for Switzerland”
Rules for Manufacturers from EU/EEA
Manufacturers placed in the EU/EEA also fall under this obligation to appoint their Swiss AR (called CH-REP or Swiss Rep or SAR). It is least expected that a single CH-REP is appointed for devices that belong to the same medical device group or family.
What does “In a document accompanying the device” mean and where should I make this information available?
This information can be affixed to the device or be available separate from the device. Available separate refers to documents such as delivery notes, customs documents, guarantee certificates, invoices, a sticker on the packaging or the instructions for use.
Such documents are made available with the device throughout the supply chain route. It is available until it reaches the distributors and need not be necessarily available to the end customer. Note that this accompanying interpretation differs from the Swiss with EU MDR.
Is the definition of a “document accompanying the device” of Swiss MedDO and Europe’s MDR 2017/745 the same?
No, they are not the same. The interpretation of document accompanying the device definition differs from Swiss MedDO and EU MDR.
In what languages must the medical device product information such as Labelling and Instructions for Use be written for the Swiss market?
It must be written and made available in all three official languages of Switzerland (German, French and Italian). Symbols established by using technical standards may be used to replace the written statements.
What are the other obligations of a Swiss AR / CH-REP?
A detailed obligation/responsibilities of a CH-REP are available in our article Obligations of the Swiss Authorised Representative.
Who are the Economic Operators required to be registered with Swiss medic?
Manufacturers (or) their Swiss Authorised Representative must register themselves within three months of placing the device for the first time in the Swiss market.
During registration, they must cater information as per Part A of Annex VI EU MDR 2017/745. Further obligations and registration modalities are governed by Article 30 and Article 31 of EU MDR 2017/745
1. Manufacturers outside Switzerland will need to appoint Swiss AR.
2. The Manufacturer will use commercially reasonable efforts to update its technical
documentation for the devices, to comply with the requirements of the ordinance, and to complete a conformity assessment procedure for devices in due time prior to the expiration of their current certificate of conformity.
3. The Manufacturer reserves the right but needs to inform AR about any discontinuities of the devices upon expiration of their current certificate of conformity.
4. As per the latest application of the ordinance, for all devices made available on the Swiss market, the manufacturer should ensure:
I. The device should be CE marked.
II. The technical documentation and the declaration of conformity of the device has been drawn up.
III. Requesting Swissmedic for the Swiss Single Registration Number (CHRN), which is used to identify the manufacturer, authorised representative, or importer and is will also need to register in the EUDAMED database issuing the Single Registration Number (SRN).
IV. Labelling of the device in accordance with the ordinance accompanied by the required Instructions For Use (IFU).
V. Assigning UDI to medical devices before placing them in the market and reporting to Swissmedic.
VI. Establishment of a post-market monitoring system and defined necessary corrective measures.
VII. Maintenance of the quality management system.
5. Manufacturers should report serious incidents associated with their medical devices to Swissmedic and should also initiate a Field Safety Corrective Action (FSCA) monitored by Swissmedic for the medical devices placed on the market in Switzerland. FSCA is used to reduce risk of death or severe deterioration in the health of the end-user associated with the medical device and may include the return of a medical device to the supplier, device – modification, exchange, or destruction.
6. The Manufacturer will not delegate to the AR the obligations laid down in Articles 16 – 17, 46 – 47, 50, 56, and 66 of the ordinance. The corresponding obligations in the (EU) 2017/745 are Article 10 (1) – (4), (6) – (7) and (9) – (12).
Obligations of the Switzerland AR Responsibilities
1. Verify the DoC and technical documentation have been drawn up and where necessary conformity assessment procedure has been carried out.
2. Keep available a copy of the technical documentation, the declaration of conformity and, if applicable, a copy of the relevant certificate, including any amendments and supplements, at the disposal of competent authorities for a period as defined in the section «Records» of this agreement.
3. Register itself on the electronic system «EUDAMED» and obtain the Single Registration Number (SRN)
4. Verify that the manufacturer has complied with the registration obligations in EUDAMED by entering the UDI core data elements (as per Article 27 of (EU) 2017/745) and has assigned and entered in Eudamed the Basic UDI-DI Information of the devices (as per Article 29 of (EU) 2017/745).
5. In response to a request from the competent authority, provide the competent authority with all the information and documentation necessary to demonstrate the conformity of a device, in an official language determined by the competent authority.
6. Forward to the manufacturer any request by a competent authority for samples, or access to a device and verify that the competent authority receives the samples or is given access to the device.
7. Cooperate with competent authority on any preventive or corrective action taken to eliminate or, if that is not possible, mitigate the risks posed by devices.
8. The AR is equally liable for any defective device in the Swiss market when either of the parties does not fulfil their obligations in accordance with the Ordinance. Therefore, it is important to consider the inclusion of AR in the corresponding processes and management reviews as deemed necessary.
9. Immediately inform the manufacturer about complaints and reports from healthcare professionals, patients and users about suspected incidents related to a device.
10. AR shall ensure that it has permanently and continuously at its disposal at least one the person responsible for regulatory compliance who possesses the required expertise (as per article 52 of MedDO / article 15 of (EU) 2017/745) regarding the regulatory requirements for medical devices in Switzerland. The PRRC shall comply with its responsibilities as defined in the Ordinance.
11. The PRRC of AR is responsible to verify the quality functions of the manufacturer (if conformity of the device is checked as per the QMS, the PMS obligations, reporting obligation of incidents, issuance of the signed statement confirming the safety of the investigational device). This could be facilitated by allowing the PRRC of AR to have access to the QMS documentation. For example, the PRRC of AR could be part of the internal audit of the manufacturer.
12. The AR is legally liable for defective devices on the Swiss market on the same basis as, and jointly and severally with, the manufacturer when the manufacturer has not complied with its obligations according to article 10 of (EU) 2017/745
1. Who is responsible for medical device regulation in Switzerland?
The Swiss Agency regulates medical devices for Therapeutic Products (Swiss medic)
2. Which current Switzerland medical device regulations need to be followed by stakeholders?
From May 26th, 2021, Stakeholders need to follow the revised Medical Devices Ordinance. (MedDO) and a new Ordinance on Clinical Trials with Medical Devices (CTO-Medd).
3. What is the EU- Switzerland mutual recognition agreement (MRA)?
EU- Switzerland mutual recognition agreement (MRA) is a crucial agreement between the EU and Switzerland. It allows the bilateral trade of several goods like medical devices, heavy machines, vehicles, etc. This MRA allowed mutual recognition of conformity assessment certificates between Europe and Switzerland.
4. What is the status of the EU-Switzerland mutual recognition agreement (MRA) for medical devices?
MRA is not updated on May 26th, 2021. As a result, the new medical device regulation of the European Union, i.e., EU MDR 2017/745, is not included in MRA. MRA has previous directives 90/385/EEC and 93/42/EEC in the medical chapter.
5. What are the consequences of this MRA status on the Swiss manufacturers/authorised representative who wants to place their products on the EU market?
From now on, Switzerland will be treated like a third-world country by the European Union. Manufacturers & authorised representatives based in Switzerland will need to appoint an EU Authorised representative in the EU and followed EU-MDR 207/745 to place medical devices on the EU market. Medical devices will need to undergo conformity assessment procedures by Notified Bodies in the EU to get CE certificates. All the existing CE certificates based on directives 90/385/EEC and 93/42/EEC under the MRA will not be considered valid in the European Union.
6. Which regulations do I need to follow in order to place my device on the swiss market?
Manufacturers will need to follow the revised Medical Devices Ordinance (MedDO) to place their products in Switzerland.
7. What are MedDO requirements?
Primary requirements of MedDO include the appointment of Switzerland authorised representative (Switzerland AR) for manufacturers based outside of Switzerland, economic operators registration and continuous reporting of severe incidents to Swissmedic, UDI assignment, and device notification.
8. Who needs to register with Swissmedic?
Economic operators (manufacturers, importers, authorised representatives placed in Switzerland) need to register themselves with Swissmedic in the first three months of
placing the device in the market. After successful registration, they will be assigned a Swiss Single Registration Number (CHRN). The Swiss Single Registration Number (CHRN) is a unique identification number to identify. Economic operators who already sell products before May 26th, 2021, in accordance with MDR 2017/745 & IVDR 2017/ 746, registrations in accordance must be completed by November 26th, 2021.
9. What is the process of economic operator registration with Swissmedic? What are the registration fees?
The economics operator needs to submit the information to Swissmedic with the help of a registration form: Application registration CHRN. Swissmedic will review the information. Once it is successfully reviewed, the economic operator will receive CHRN. For the assignment of CHRN, fees will be based on the amount of review work done by Swissmedic per application. It is CHF 200 per hour.
10. What Information is required by Swissmedic for economic operators registration?
Name of the registrant, contact details, details of the person responsible for regulatory compliance, commercial Register extract, For Swiss-domiciled, authorised representatives to mandate from a manufacturer outside Switzerland is required by Swissmedic.
11. Who can be appointed as Switzerland AR? What is the deadline for appointing Switzerland AR?
A Swiss authorized representative (Swiss AR) is a legal person that needs to be appointed by a manufacturer established outside of Switzerland to place the products on the Swiss market. A written mandate/letter designating an authorised representative for a medical device manufacturer is mandatory. Any legal entity or natural person situated in Switzerland can act as Switzerland AR. Switzerland AR is responsible for product safety and will be held liable for product defects. Manufacturers can appoint only a single Authorised representative in Switzerland.
Timeline to appoint Switzerland AR:
12. Which devices need to be notified to Swissmedic? What is the process?
The following device needs to be notified to Swissmedic before placing it on market:
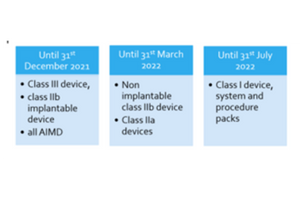
13. What is the timeline to assign UDI? For placing UDI- carriers on the labels of the device deadline for implantable devices and
class III devices are 26th May 2021, Class IIa and class IIb devices are 26th May 2023 and for
class I devises it is 26th May 2025.
14. Will the Swissmedic accept CE certificates? CE certificates from notified bodies placed in EU/EEA countries will be valid in Switzerland until and unless the applied conformity assessment procedures comply with the Swiss requirements meet, and they are issued by a notified body that has an equivalent qualification as described in MedDO.
15. What are the regulations for the In vitro diagnostic devices? In vitro, diagnostic medical devices regulations are in the final step. They will be published as a separate ordinance on in-vitro diagnostics (IvDO).