SaMD is software intended for one or more medical purposes that perform these without being part of a hardware medical device.
Medical device software is meant to be used, alone or in combination, for a purpose specified in the definition of a “medical device” in the MDR or IVDR, regardless of whether the software is independent or driving or influencing the use of a device.
To be qualified as medical device software, the product must first fulfil the software definition according to this guidance and the description of a medical device according to Article 2(1) of Regulation (EU) 2017/745 – MDR.
To be qualified as an in vitro diagnostic medical device software, the product must additionally fulfil the definition of an in vitro diagnostic medical device according to Article 2(2) of Regulation (EU) 2017/746 – IVDR.
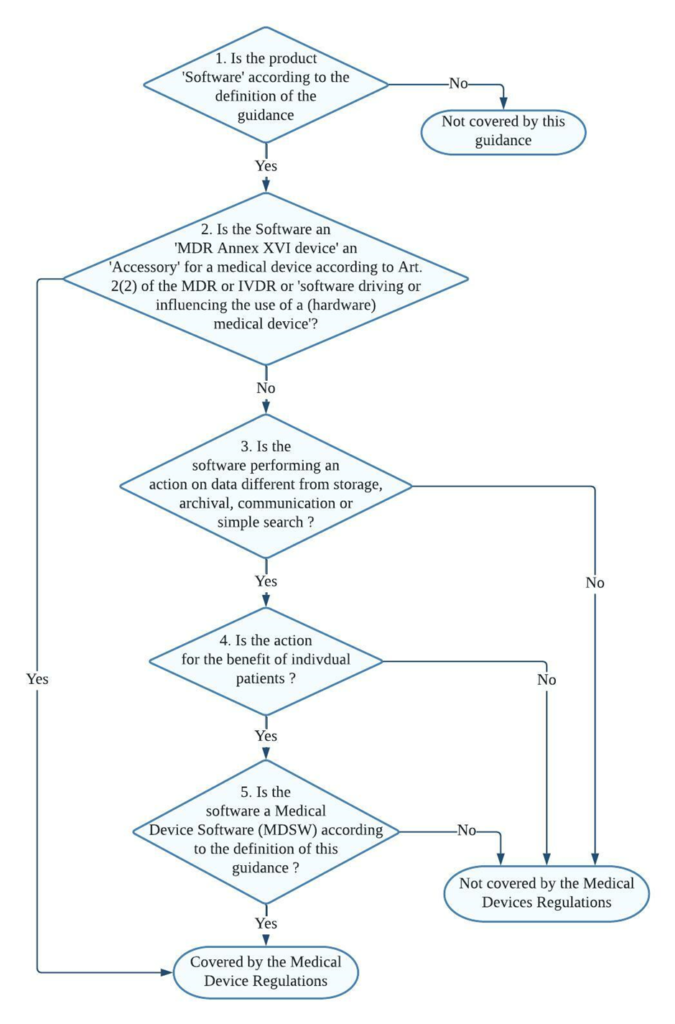
Decision steps for qualification of software as MDSW
Decision step 1: If the product is software according to Section 2 of the guidance (link provided at the bottom), then it may be a medical device software; proceed to decision step 2; else, it is not covered by this guidance but may still be covered by the Medical Devices Regulations.
Decision step 2: If the product is an MDR Annex XVI device or an accessory for a medical device, or is software driving or influencing the use of a medical device, then it must be considered as part of that device in its regulatory process or independently if it is an accessory. If it is not, proceed to decision step 3.
Decision step 3: if the software does perform an action on data or performs an action beyond storage, archival, communication, simple search, lossless compression, then it may be a medical device software; proceed to step 4.
Decision step 4: is the action for the benefit of individual patients?
Examples of software that are not considered as being for the use of individual patients are those which are intended only to aggregate population data, provide generic diagnostic or treatment pathways (not directed to individual patients), scientific literature, medical atlases, models, and templates as well as software intended only for epidemiological studies or registers.
Decision step 5: Is the software medical device software (MDSW) according to the definition of this guidance?
Decision steps for qualification of MDSW as either a medical device or an in vitro diagnostic medical device
Decision Step 1: Does the Medical Device Software (MDSW) provide information within the scope of the in vitro diagnostic medical device definition?
MDSW, which provides information according to Regulation (EU) 2017/746 – IVDR Article 2(2), should qualify as In Vitro Diagnostic Medical Device Software (IVD MDSW)
Concerning a physiological or pathological process or state (by investigation of this process or state)
Concerning congenital physical or mental impairments
Concerning the predisposition to a medical condition or a disease
To determine the safety and compatibility with potential recipients
To predict treatment response or reactions
To define or monitor therapeutic measures.
An MDSW that falls under the definition set out in EU Article 2 (1) of Regulation (EU) 2017/745 – MDR should qualify as Medical Device Software (MD MDSW). In specific, the following considerations should apply to the provision of information by software:
Diagnosis, prevention, monitoring, prediction, prognosis, treatment or alleviation of disease
Diagnosis, monitoring, treatment, alleviation of, or compensation for, an injury or disability
The investigation, replacement, or modification of the anatomy or a physiological or pathological process or state
Control or support of conception
Products specifically intended for the cleaning, disinfection or sterilisation of devices as referred to in Article 1(4) and Annex XVI products.
Decision Step 2: Does the MDSW create information based on data obtained by in vitro diagnostic medical devices only?
Suppose the information provided is based on data obtained solely from in vitro diagnostic medical devices. In that case, the software is an in vitro diagnostic medical device and is, therefore, an IVD MDSW.
If the data analysed is obtained from both in vitro diagnostic and medical devices, proceed to step 3.
Decision Step 3: Is the intended purpose substantially driven by in vitro diagnostic medical devices?
The applicable legislation is Regulation (EU) 2017/746. If the intended purpose is substantially driven by data sources coming from medical devices, then the relevant legislation is Regulation (EU) 2017/745.
In the condition where the intended purpose of the MDSW output data fulfils both the medical device and in vitro diagnostic medical device definitions set out in the MDR and IVDR, a weighting of the data sources based on the significance of the information concerning fulfilling the intended purpose should be conducted to aid the manufacturer in determining which regulation to apply.
Consideration of changes to an MDSW
Manufacturers shall evaluate the potential impact of any changes to the function, intended use, basic design, and manufacturing characteristics on the software’s qualification as MDSW and its classification.
It is to be noted that a change to or the addition of functionality to software may lead it to be qualified as MDSW or a revision of the classification of the MDSW. Similarly, a module that is added to software might be equipped as a MDSW on its own.
When determining the risk class of a combination of a modified MDSW and a medical device, the intended purpose and functionality of that (new) combination must be considered.
FAQs
How MDD considered SaMD classification approach and what difference has MDR brought in it ?
Most standalone medical software falls into the lowest risk category — Class I — under the EU MDD. The only software used for specialised diagnostic activities obtained a higher classification.
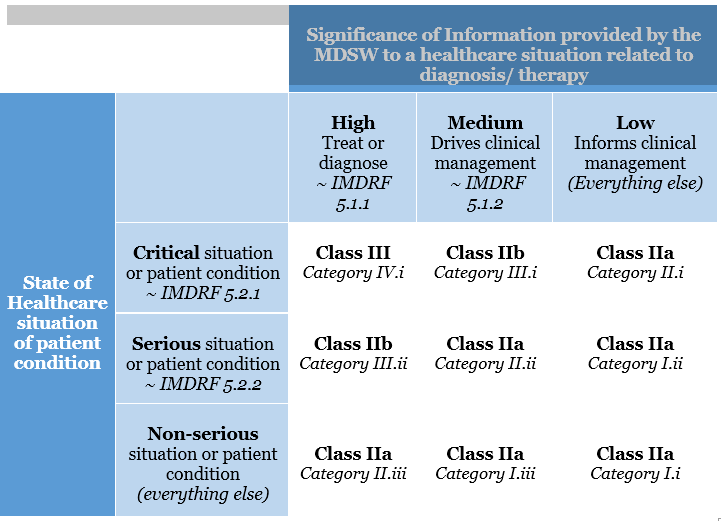
This categorisation has been altered in the EU MDR. The risk classification of software is covered under Rule 11. The legislation specifies that software used for diagnostic or therapeutic purposes must be classified as Class IIa (or higher). Rule 11 was added to the MDR to address the risks associated with information provided by an active device, such as the MDSW. The significance of the information provided by the active device to the healthcare decision (patient management) in connection with the healthcare situation is described and categorised in Rule 11.
Rule 11 states:
Software intended to provide information that is used to make decisions with diagnosis or therapeutic purposes is classified as class IIa, except if such decisions have an impact that may cause:
Death or an irreversible deterioration of a person’s state of health, in which case it is in class III
Severe deterioration of a person’s state of health or surgical intervention, in which case it is classified as class IIb.
Software intended to monitor physiological processes is classified as class IIa, except if it is designed for monitoring vital physiological parameters, where the nature of variations of those parameters is such that it could result in immediate danger to the patient, in which case it is classified as class IIb.
All other software is classified as class I.
What is the transition plan from MDD to MDR for SaMD devices ?
While many devices approved under the MDD can continue to be sold until 2024, Class I items and those that have been “up-classified” due to the MDR do not have that opportunity; they must be compliant by May 2020.
The MDR regulation’s wording has been clarified – it’s all about the expiration dates of CE mark certificates. MDD-certified products can be used until the expiration date on their CE-marking certificate (a soft transition, potentially up to 2024). However, there are specific additional requirements, such as post-market surveillance. On the other hand, Class I products are self-certified and so lack a certificate with an expiration date. At the end of the transition period, they must be compliant on May 26, 2020.
How is MDSW classified under IVDR ?
In determining the proper classification of MDSW under the IVDR, the manufacturer shall consider all categories and implement the rules of Annex VIII of the IVD Regulation (EU) 2017/746.
Examples for the classification of MDSW under the IVDR:
Software intended to be installed on a fully automated enzyme-linked immunosorbent assay (ELISA) analyser, and intended to determine the Human HbA1c concentration in serum from the results obtained with a Human HbA1c ELISA, designed to screen for and diagnose diabetes and monitor diabetic patients, should be in class C per Rule 3(k).
Software within a PAP stain automated cervical cytology screening system intended to classify the PAP cervical smear as usual or suspicious should be in class C per Rule 3(h).
Software for the interpretation of automated readings of line immunoassay to confirm and determine antibodies to HIV-1, HIV-1 group O and HIV-2 in human serum and plasma should be in class D per Rule 1.
Software that uses maternal parameters such as age, the concentration of serum markers and information obtained through foetal ultrasound examination for evaluating the risk of trisomy 21 should be in class C per Rule 3(l).
Classification examples in Annex IV are provided for guidance purposes and illustrate how a particular rule may be applied to a device. The indicated classification in the example is not a confirmation of the final classification of the device, as other laws must also be considered.
What are the risks SaMD medical devices possess ?
While SaMD has the potential to improve the healthcare system significantly, the International Medical Device Regulators Forum (IMDRF), a collection of medical device regulators working toward international regulatory harmonisation, has recognised that SaMD poses new problems to medical device regulation.
Regulators face the difficult task of regulating software that undergoes frequent changes, potentially affecting the software’s safety and efficacy. Because the conventional physical constraints of containment are no longer present, the ability to download the software through the internet creates a risk.
To identify and track SaMD throughout its life, regulators and the industry must agree on globally applicable device identification and coding standard. They’re not like a standard medical device, which can be labelled with a Unique Device Identifier on the outside.
Another issue with SaMD is the software’s security. The medical device’s use may be jeopardised due to cybersecurity vulnerabilities. It could allow the attacker to take control of the device remotely, modify its operation and compromise its safety or effectiveness, or expose confidential data.
From a regulatory standpoint, the following are some of SaMD’s challenges:
Medical Device Regulations establish guidelines for categorising medical devices from low to high risk. Some SaMD do not fall neatly into the classification scheme designed for traditional medical devices.
Artificial Intelligence is already improving several SaMD systems in image-based healthcare by continuously learning from the data provided to the software.
MSDW Risk Classification based on Rule 11 of the MDR
Clinical investigation creates a set of clinical data that is crucial to understanding the effectiveness of a medical device. Clinical Investigation is done for devices that are previously UKCA / CE / CE UKNI marked or for research medical devices.
It is governed by a clinical investigation plan with an objective, methodology, and record keeping.
Steps involved in Clinical Investigation in UKCA
Step 1:
Ensure all the information necessary to demonstrate compliance with all the relevant essential requirements is available. Appoint an Authorised representative/Responsible person.
This representative communicates to the Medicines and Healthcare products Regulatory Agency (MHRA) on behalf of Manufacturers.
Step 2:
Please provide the MHRA with advanced notice of your intention to submit a clinical investigation by emailing [email protected] with some basic details about the investigational device, the intended population, the type of study, and the estimated application date.
They conducted in Great Britain must follow UKCA Medical Devices Regulations MDR 2002. Clinical Investigations in Northern Ireland must meet the requirements of the EU MDR and be submitted to MHRA.
Complete the CI Application form and submit the list of documents. Submission of documents should be in English. If the documents are in other languages, translation into English should be made available. Supported format PDF only.
List of Documents required
• Cover letter on headed paper
• Clinical investigation plan or CIP
• Investigator’s brochure
• Participant information sheet
• Consent form from participants
• CVs of UKCA clinical investigators
• Details of the device
• General Safety and Performance Requirements (GSPR) or Essential requirements checklist
• Biological safety assessments of patient contact materials (if any)
• Information on animal tissues (if any)
• Information of any medicine or human blood derivative, or non-viable human tissues and cells incorporated into the device
• Research ethics committee opinion (if available)
Step 4:
Payment of Fees
Devices are categorised according to risk as a Group A or B device: Group A includes class I, IIa, and IIb devices other than implantable/long term invasive. Group B has class IIb implantable/long term invasive, class III, active implantable devices.
MHRA fees and guidance on making a payment for the application of clinical investigation are available on the MHRA website.
Step 5
The review process by MHRA /Assessment procedure.
When the MHRA has validated the documents, within five working days, a response is obtained. It will confirm that the 60-day assessment has started or response to issues.
If issues are raised, the 60-day evaluation will start when MHRA receives a valid response. Day 1 of the 60 days is the first working day that follows the date of receipt of a valid Notification.
Experts will check the safety and performance of your device. The MHRA will write to you if they require further information. A letter will be sent on the 60th day with a decision (‘objection’ or ‘no objection’) if the clinical investigation can be carried out or not.
If any severe events occur during the assessment, they should be reported using the MEDDEV 2.7/3 or MDCG 2020-10/2 Reporting Table.
Step 6
Notification of Amendments (if any)
Once a letter of no objection is received after the review process, you must notify the MHRA of proposed amendments to the investigation. You must wait until another letter of no objection before implementing the changes.
Any changes made to the following must be notified:
the device under investigation
study documentation, including the clinical investigation plan
investigators or investigating institutions
changes requested by an ethics committee
The following document must be presented for notification of amendments.
covering letter with:
Reference number allotted by MHRA for the CI and a table with a summary of each proposed change with the reason for the individual changes.
FAQs
Is the procedure the same for Northern Ireland and Great Britain?
The overall procedure remains the same, but the documents required may vary. This is due to the regulations followed in both regions. Northern Ireland follows EU Medical Devices Regulations 2017/745, whereas Great Britain follows UK MDR (Medical Devices Regulations) 2002.
What happens if amendments are not notified to MHRA?
All modifications should be notified to MHRA. However, only some are substantial. Manufacturers/Sponsors must notify else; they are liable to prosecution. More details can be found here.
Summary of Safety and Clinical Performance (SSCP) acts as a vital document that allows the public to access information quickly. The information in the SSCP can be sourced entirely from the technical file.
The technical file consists of the Post Market Surveillance (PMS), risk assessment, post-market clinical follow-up (PMCF) plans and reports. The SSCP document is required for high-risk devices only-this includes Class III and all implantable devices.
Manufacturers of custom-made or investigational devices need not produce this document. Implant card together with SSCP enables an efficient system to access device information.
SSCP for medical devices under MDR
Under MDD, the information on medical devices was not easily attainable. Therefore, the end-users of most medical devices were deprived of information regarding even high-risk medical devices.
As a result, medical devices under the directives lacked clarity. The main reason why SSCP is introduced is that MDR, unlike the directives, brings accessibility of information into account.
The SSCP should be made available on the EUDAMED website for easy access. It should be assigned an identifier that remains the same throughout the document’s lifetime. This identifier is not subjected to changes even when the content of this document is revised.
Language and readability requirements
SSCP follows the MDR language requirements like the other technical documents, such as Instructions for Use. All intended users within the EU understand no single language.
Therefore, language translations should be made available to intended users and patients. The SSCP should be translated into the languages accepted in the Member States where the device is intended to be sold.
Healthcare professionals widely understand English. Having an English translation available is essential, even if it is not available in selecting the official languages of each Member State. This enables the access to information that EU MDR strives to achieve.
While preparing SSCP, readability is an essential factor. The manufacturers must bear in mind to produce two sections. One part for intended users/healthcare professionals, and a second part for patients if applicable.
It is recommended to use an appropriate method to confirm that the document is understandable to the member of both categories. Further guidance on the readability, translations and other factors involved in SSCP can be found in the MDCG guidance on Summary of safety and clinical performance.
Sections of Summary of Safety and Clinical Performance
Article 32 of the EU MDR states some sections that are a must. The manufacturer may add further information from the Technical Dossier/File if relevant to the users.
The following sections are mandatory in an SSCP for healthcare professionals:
Details of the device and manufacturer (including UDI details).
Intended use of the device.
Description of the device and its components. Previous variants of the device.
Indications, contraindications, and target demographic.
Details of residual risks, undesirable effects, warnings, and precautions
Risks, undesirable effects, and warnings should be mentioned in SSCP. Other relevant aspects of safety, serious events, and a summary of any field safety corrective action must be included if applicable.
A summary of the Clinical Evaluation of the device.
This section is intended to summarise the clinical evaluation results and the clinical data, the evaluation of undesirable side-effects, and the benefit-risk ratio’s acceptability.
It is an objective and balanced summary of the clinical evaluation results of all the available clinical data related to the device. It should comprise favourable, unfavourable, and inconclusive data.
Conclusions based on evidence and the safety and performance of the device.
Suggested profile and training for users
Applied harmonised standards
All commonly applied specifications and international standards harmonised and adopted monographs should be listed in SSCP.
Revision history
Revision history should contain details such as revision validated by Notified Body (NB) and the language of SSCP validated.
A patient-specific SSCP template follows the same high-level structure as the clinician SSCP but does not include the reference to harmonized standards and common specifications. This decision focused on the information most relevant to patient health.
Validation of SSCP
When the Notified Bodies (NB) have assessed that all the required elements are included in the draft SSCP with the most current version of relevant documents in the TD, the SSCP has been validated by the NB.
SSCP validation may depend on the class of device and the conformity assessment routes chosen. More guidance on validation by NB can be found in the MDCG guidance on SSCP.
Uploading SSCP to EUDAMED
SSCP should be made available online for the users who intend to read the document. It is uploaded in EUDAMED by the Notified Bodies, the only actor managing the SSCPs in EUDAMED.
After each validation process, NB shall upload the updated SSCP by replacing the older version. However, once the ‘master’ SSCP is uploaded, it is up to the manufacturer to upload the translations to EUDAMED.
FAQs
What resources can be used for the SSCP?
The technical files like the design validation report, risk management report, clinical evaluation report (CER), as well as Post-Market Surveillance (PMS) and Post-Market Clinical Follow-up (PMCF) reports.
The Instructions for Use of the device can also be used as information for preparing the SSCP. Please note that SSCP cannot replace the IFU.
How frequently should SSCP be reviewed or updated?
The SSCP should be ideally updated annually. Documents like the PMCF evaluation and periodic safety update reports (PSUR) are updated annually. It is recommended to update SSCP along with the other technical documents.
How can I access the SSCP?
SSCP will be made available with the launch of the EUDAMED database. SSCP is accessible to both healthcare professionals and patients. SSCP has clear information for healthcare professionals with prior knowledge of medical terminologies.
In addition to this, SSCP should also contain a section that patients of different levels of expertise easily understand.
When should a manufacturer of Class III medical devices prepare SSCP?
SSCP should be made available at the time of registration.
Is there a similar requirement under IVDR?
Article 29 of In Vitro Diagnostics Regulation 2017/746 (EU IVDR) describes a requirement like the SSCP: the Summary of Safety and Performance (SSP).
The SSP requirements are almost identical to those of the SSP, replacing ‘clinical’ for ‘performance’ evaluation. Also, it includes a requirement for metrological traceability of assigned values intended for analytes used in IVDs.
For more information on IVDR, read our article on IVDR 2017/746.
Under both the EU MDR and the EU IVDR, the MDCG rules specify the conditions for a product to qualify as medical device software (MDSW). An MDSW product can enter the market in one of the ways:
As a standalone medical device or
As a component of another medical device regulation.
The former demands a lengthy regulatory process that includes a conformity assessment to determine whether the medical device meets EU MDR requirements. However, that rule does not apply to products in the latter group with the same rigour.
As a result, MDSW, as an integral component of a medical device, can be placed on the market through the medical device’s conformity assessment route.
The term “Software as a Medical Device” (SaMD) is defined as Software intended to be used for one or more medical purposes that perform the below purposes without being part of a hardware medical device.
SaMD is a medical device and includes an in-vitro diagnostic (IVD) medical device regulation.
SaMD is capable of running on general-purpose (nonmedical purpose) computing platforms.
“Without being part of” means Software is not necessary for a hardware medical device regulation to achieve its intended purpose.
The Software does not meet the definition of SaMD if its intended purpose is to drive a hardware medical device regulation.
Examples of Software as a Medical Device
SaMD may be interfaced with other medical devices, including hardware medical devices and other Software such as medical device software and general-purpose Software. The Software provides parameters that become the input for a different hardware medical device or other SaMD. For example, treatment planning software that supplies information used in a linear accelerator is Software as a Medical Device.
Software with a medical purpose that operates on a general-purpose computing platform, i.e., a computing platform that does not have a medical purpose, is considered Software as a Medical Device. For example, Software intended to diagnose a condition using the tri-axial accelerometer that operates on the embedded processor on a consumer digital camera is considered Software as a Medical Device.
Software that is connected to a hardware medical device but is not needed by that hardware medical device regulation to achieve its intended medical purpose is Software as a Medical Device and not an accessory to the hardware medical device regulation.
Software as a Medical Device can run on general-purpose (nonmedical purpose) computing platforms.
Top 7 Manufacturer Obligations for Medical Device Regulation
For SaMD manufacturers, the definition in GHTF/SG1/N55:2009 applies: “Manufacturer” means any natural or legal person with responsibility for the design and manufacture of a medical device to make the medical device available for use under his name; whether such a medical device is designed and manufactured by himself or on his behalf by another person(s).
Unless the Regulatory Authority (RA) within that jurisdiction imposes this responsibility explicitly on another person, this ‘natural or legal person’ has ultimate legal responsibility for ensuring compliance with all applicable regulatory requirements for the medical device in the countries or jurisdictions where it is intended to be made available or sold.
The manufacturer’s responsibilities are described in other GHTF guidance documents. These responsibilities include meeting pre-market and post-market requirements, such as adverse event reporting and notification of corrective actions.
‘Design or manufacture’, as referred to in the above definition, may include specification development, production, fabrication, assembly, processing, packaging, repackaging, labelling, relabelling, sterilisation, installation, or remanufacturing of a medical device; or putting a collection of devices, and other products, together for a medical purpose.
Any person who assembles or adapts a medical device already supplied by another person for an individual patient, per the instructions for use, is not the manufacturer, provided the assembly or adaptation does not change the intended use of the medical device.
Any person who changes the intended use of, or modifies, a medical device regulation without acting on behalf of the original manufacturer and who makes it available under his name should be considered the manufacturer of the limited medical device.
An authorised representative, distributor or importer who only adds its address and contact details to the medical device or the packaging without covering or changing the existing labelling is not considered a manufacturer.
To the extent that an accessory is subject to the regulatory requirements of a medical device, the person responsible for the design or manufacture of that accessory is considered to be a manufacturer.
Quality Management Principles
Medical device QMS principles allow the measurement of activities depending on
The type of medical device.
Risk of the product to patients.
Size of the organisation.
Technology or automation is used to manufacture.
And other factors are determined by the manufacturer to control quality and maintain the safe and effective performance of the medical device regulation.
The manufacturing of SaMD, a software-only product, is primarily based on the development lifecycle activities often supported by automated software development tools.
These computerised activities may, in some cases, replace discrete or deliberate actions (e.g., transfer of design to production) typically found in the manufacturing of hardware products.
However, the principles in a QMS that provide structure and support to the lifecycle processes and activities are still applicable and essential to controlling the quality of SaMD.
An effective QMS for SaMD have to include the following principles:
An organisational structure ensures SaMD’s safety, efficacy, and performance by providing leadership, accountability, and governance with enough resources.
A set of SaMD lifecycle support processes that are scalable to the organisation’s size and applied consistently across all realisations and use processes
A set of realisations and use processes that are scalable for the type of SaMD and the organisation’s size takes into account essential elements required for assuring the safety, effectiveness, and performance of SaMD.
FAQs
Does MDSW need to be re-assessed for risk classification when transitioning from MDD to MDR?
Yes, SaMD also requires a re-assessment of risk classification since the new classification rules are more stringent than the MDD. This must be carried out as the first step in the MDR compliance roadmap.
The gap analysis in the technical documentation also needs to be performed while transitioning to MDR.
What are the additional requirements in the technical documentation for MDSW in MDR?
Pre-clinical & Clinical Data, Clinical Evaluation and PMCF requirements, Clinical Investigation and implementation of UDI.
How is Software as a Medical Device considered in MDR?
Software shall be deemed to be an “active device” both in MDR and IVDR.
What are the UDI assignment criteria for a Software only device?
For a Software that constitutes as a device on its own, the UDI shall be assigned at the system level of the Software itself. The UDI that is applied to the physical medium that contains the Software shall be identical to the UDI assigned at the system level.
Are there any guidance documents for Software as a Medical Device by the EU commission?
Yes, Specifically
“MDCG 2019-16 Guidance on Cybersecurity for medical devices” guides manufacturers on how to fulfil the Annex I requirements regarding cybersecurity.
“MDCG 2019-11 Guidance on Qualification and Classification of Software” in Regulation to MDR 2017/745 and IVDR 2017/746
The classification of medical devices in the UK market will be based on UK MDR 2002 from 1st January 2021. Medical devices are classified into a particular class based on the level of risk they pose to the patient and according to the intended purpose of use.
The medical devices are classified based on the classification rules in Annex IX of Directive 93/42 into one of the following four classes:
Class I – Medical device which poses low to moderate risk to the patient
Class IIa – Medical device which poses a moderate risk to the patient
Class IIb – Medical device which poses a moderate to high risk to the patient
Class III – Medical device which poses a high risk to the patient
The medical devices are classified into the above-given classes based on 18 rules mentioned in Annex IX of Directive 93/42.
These 18 rules are majorly divided into four parts which are non-invasive devices, invasive devices, active devices and special rules.
Devices that do not penetrate inside the body fall under non-invasive
Devices that penetrate inside the human body either through a body orifice or through the surface of the body are Invasive devices (this also includes surgically invasive devices)
Devices that require an external energy source come under active devices
All other devices falling outside of these categories come under Special rules
In the event of a dispute between a manufacturer and a notified body over the classification of a device, the matter shall be referred to the Secretary of State, who shall determine the classification of the device in accordance with the classification criteria set out in Annex IX of Directive 93/42.
Few Common examples of the various classes discussed above:
Class I – Bandage, Spectacle Frames, Surgical Masks, Wheelchair
Class IIa – Suture Needles, Hearing Aids, Surgical clamps
Class IIb – Infusion pumps, Surgical Laser, Ventilator
Class III – Pacemaker, defibrillator, Cochlear implants
Rules to Remember for UK MDR 2002
Medical devices which are used in a particular combination are also individually classified into their specific class
Software that is used to aid the operation of a particular medical device also falls into the same class as the medical device
The medical device that is not explicitly designed for a particular part of the body is be classified based on the most critical use of that device
If multiple rules apply to a particular medical device, the rule which involves the highest risk must be considered for the classification purpose
4 General Procedure of Medical Devices Based on UK MDR 2002
Decide the type of device that you are about to classify into a non-invasive, invasive, active, or special category.
Look at each UK MDR 2002 classification rule and determine which rule applies to your medical device.
If multiple rules apply for your device apply the highest risk rule.
If the product is combined determine its principal mode of action with respect to its intended purpose and apply the most suitable rule.
Once the medical device is successfully classified into a particular class, the registration process or the conformity assessment for that medical device will be based on the derived risk class of the device. Hence Classification of the medical device plays an integral role in the registration process of a medical device
FAQs
For an integrated medical device that contains both hardware and software, with software being part of the entire functionality of the device, what is the basis of classification?
For an integrated medical device, the classification of the device is based on the risk imposed by the medical device in terms of safety. Classification of the software is A, B or C based on which the approach of software life cycle processes is carried out.
However, as an integrated medical device, it is classification is I, II, III & IV depending on the risk impact to the patient.
What if my medical device is a Drug-Device Combination or contains a Medicinal Product?
Where a device is intended to administer a medicinal product within the meaning of Article 1 of Directive 2001/83/EC, that device shall be governed by this Directive, without prejudice to the provisions of Directive 2001/83/EC with regard to the medicinal product.
If, however, such a device is placed on the market in such a way that the device and the medicinal product form a single integral product which is intended exclusively for use in the given combination and which is not reusable, that single product shall be governed by Directive 2001/83/EC.
The relevant essential requirements of Annex I to this Directive (93/42) shall apply as far as safety and performance-related device features are concerned.
What are the new devices that are to be classified other than the medical device?
Accessories for medical devices can be separately classified in their own rights of use, independent of the classification applied to the main medical device.
Products that are used in combination with other product(s), the classification rule applies separately to each individual product.
Software, which drives a device or influences the use of a device, falls automatically in the same class.
Will the Quality Management System requirements of EU MDR be in effect for the UK in future?
At this moment, the UK MDR 2002 has adopted the EU Directive 93/42. Nevertheless, manufacturers are expected to stage up their QMS processes for reviewing their pre and post-market regulatory procedures, clinical evidence, and risk management.
Performing a gap assessment is significant to understand and prepare the road map to accelerate towards compliance for the regulations.
On 5 May 2017, the EU published the new EU MDR 2017/745 and IVDR 2017/746 regulations in which they formally introduced the UDI system in the EU. The UDI comprises the following components
A device identifier (UDI-DI)
A production identifier (UDI-PI)
The Basic UDI-DI is a technique introduced by the EU for linking medical devices to their regulatory documentation so that the model of the product can be uniquely identified throughout its entire lifecycle.
The linked documentation may include the declaration of conformity, notified body certificates, technical documentation and a summary of the safety and clinical performance of the device.
The UDI-DI identifies specific, detailed information about a particular device. The UDI-DI is a unique numeric or alphanumeric code specific to a device model and used as the ‘access key’ to the information stored in a UDI database.
If any of the below details change, the device will then need a new UDI-DI.
Name or trade name of the device
Device version or model
Labelled as a single-use device
Packaged as sterile
Maximum number of uses
Need for sterilization before use
Quantity of devices provided in a package
Critical warnings or contra-indication
CMR/endocrine disruptors
Before placing a device for clinical or performance evaluation on the market, the manufacturer has to assign a Basic UDI-DI to the device and enter it into the UDI and Device Registration module of EUDAMED, along with other data elements related to that device.
The Basic UDI -DI should be maintained by the manufacturers by establishing processes for the assignment and maintenance of Basic UDI-DIs.
According to the new rules, any manufacturer can assign a UDI to the device and to all the higher levels of packaging of the device as well before the device is placed on the market. The label of the device should have the UDI carrier.
The manufacturer has to ensure that before the device is placed on the market, the information related to the device has to be submitted correctly and saved to the UDI database. In the EU, the manufacturer has to assign a UDI along with a Basic UDI-DI to all his devices.
Some important points on the UDI:
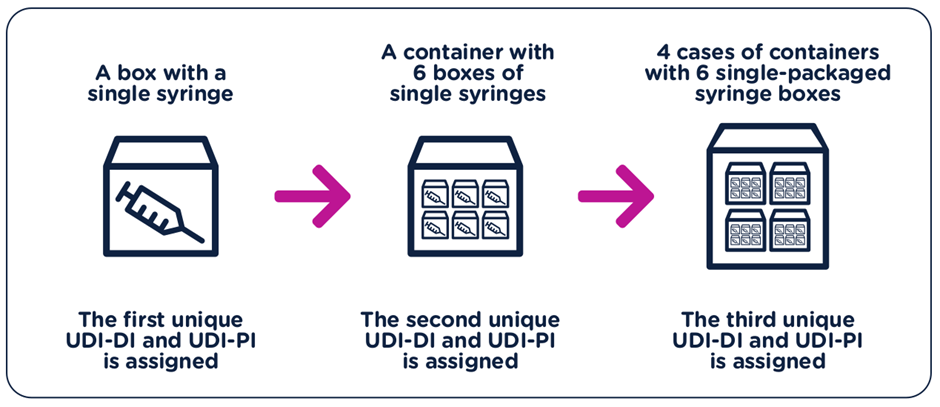
The UDI-DI have to be unique at each level of device packaging.
Each component considered to be a device and is also commercially available on its own must be assigned a separate UDI unless the components are part of a configurable device that is marked with its own UDI.
A new UDI-DI is required whenever there is a change that could lead to misidentification of the device or poses an error for its traceability.
The data for new UDI-DIs shall be available at the time the device is placed on the market.
Manufacturers shall update the relevant UDI database record within 30 days of a change being made to an element, which does not require a new UDI-DI.
Article 27 of 2017/745 and Article 24 of Regulation 746/2017 lay down that the UDI system shall consist
Of production of a UDI that comprises a UDI device identifier (‘UDI-DI’) specific to a manufacturer and a device, providing access to the information and a UDI production identifier (‘UDI-PI’) that identifies the unit of device production and, if applicable the packaged devices
Placing the UDI on the label of the device or its packaging
Storage of the UDI by economic operators, health institutions and healthcare professionals, in accordance with the conditions laid down in the article.
FAQ
What is the UDI?
The UDI is a series of numeric or alphanumeric characters created through a globally accepted device identification and coding standard.
It allows the unambiguous identification of a specific medical device on the market. The UDI is comprised of the UDI-DI and UDI-PI. The unique identifier may include information on the lot or serial number and be able to be applied anywhere in the world.
The production of a UDI comprises the following:
A UDI device identifier (‘UDI-DI’) specific to a device, providing access to the information laid down in Part B of Annex VI.
A UDI production identifier (‘UDI-PI’) identifies the unit of device production and, if applicable, the packaged devices, as specified in Part C of Annex VI.
What is the Basic UDI-DI?
The Basic UDI-DI is the primary access key for device-related information in the Eudamed database, and it is referenced in relevant documentation [e.g. certificates (including certificates of free sale), EU declarations of conformity, technical documentation, and summaries of safety and (clinical) performance]).
Its goal is to find and connect devices that have the same intended purpose, risk class, and key design and manufacturing features.
It is distinct from the device’s packaging/labelling and does not appear on any commercial item.
Any Basic UDI-DI must uniquely identify the devices (group) covered by the Basic UDI-DI.
What are the entities that provide the service of issuing UDI to medical devices?
GS1
Health Industry Business Communications Council (HIBCC)
ICCBBA
Informationsstelle für Arzneispezialitäten — IFA GmbH
How to get a Basic UDI-DI?
Basic UDI-DI should be assigned in compliance with the rules of the chosen issuing entity.
Can one Basic UDI-DI be linked with more than one UDI-DI?
Medical devices with the same intended purpose, risk class, essential design etc., are grouped under a single and unique Basic UDI-DI. As such, a medical device that has been assigned a UDI-DI can be linked to only one Basic UDI-DI. There is, therefore, a one-to-one relationship of 1 UDI-DI per 1 Basic UDI-DI. On the other hand, one Basic UDI-DI can be linked to multiple UDI-DIs. Therefore, there is a relationship of 1 Basic UDI-DI to N UDI-DIs.
Which products are subject to the UDI system?
The UDI system should apply to all devices, except custom-made and performance study/investigational devices.
Who is responsible for placing the UDI carrier on the device itself, on the label and the device’s package?
All UDI-related requirements are the responsibility of the manufacturer. This comprises assigning the UDI (and Basic UDI-DI), registering the UDI (and Basic UDI-DI) in the Eudamed database, and placing the UDI carrier on the device’s label, packaging, or, in the case of reusable devices, on the device itself.
What are the obligations of economic operators and health institutions in relation to UDI?
According to the two medical devices Regulations, manufacturers shall be responsible for the UDI assignment and placement of the UDI carrier, the initial submission and updates of the identifying information and other device data elements in the Eudamed database.
Manufacturers shall update the relevant database record within 30 days of a change being made to an element which does not require a new UDI-DI.
Distributors and importers shall verify that, where applicable, a UDI has been assigned by the manufacturer. All economic operators and health institutions shall store and keep preferably by electronic means the UDI of the devices, which they have supplied or with which they have been supplied if those devices belong to class III implantable devices.
Please note that the Commission may decide to adopt implementing acts to expand the scope of devices for which economic operators shall store and keep the UDI.
What is the procedure for systems and procedure packs to undergo a UDI registration?
Systems and procedure packs shall undergo a UDI registration, as described in MDR Article 29(2).
Before placing on the market a system or procedure pack pursuant to Article 22(1) and (3), that is not a custom-made device, the system or procedure pack producer shall assign to the system or procedure pack in compliance with the rules of the issuing entity, a Basic UDI-DI and shall provide it to the Eudamed database together with the other relevant core data elements listed in the MDCG 2018-4 guidance document.
Are devices, which are compliant with the Medical Device Directives (MDD and AIMDD) and placed on the market after the application date of the Regulations (legacy devices), continue to be subject to UDI requirements?
In order to facilitate the transition to the new system, the new regulations allow manufacturers to place products on the market after the general application dates of the new Regulations (and until 26 May 2024 at the latest) under valid Directive certificates.
These legacy devices are not subject to UDI obligations, but they should be registered in the Eudamed database.
Timelines for registration as described under question 6 also apply to these products. More information on the operational aspects of the registration of legacy devices is available in the MDCG 2019-5 guidance document.
How should a UDI appear on the label or package of a device?
The UDI Carrier [Automated Identification for Data Capture (AIDC) and human-readable interpretation (HRI) representation of the UDI] shall be on the label or on the device itself and on all higher levels of device packaging.
In the event of significant space constraints on the unit of use packaging, the UDI carrier may be placed on the next higher packaging level. Higher levels of packaging shall have their own unique UDI. Please note that shipping containers shall be exempted from the requirement.
The UDI must appear in a plain-text version/human-readable information (HRI) and in a form that uses AIDC technology. AIDC means any technology that conveys the unique device identifier or the device identifier of a device in a form that can be entered into an electronic patient record or another computer system via an automated process. The HRI consists of legible characters that people can easily read. If significant constraints limit the use of both AIDC and HRI on the label, only the AIDC format shall be required to appear on the label.
For devices intended to be used outside healthcare facilities, such as devices for home care, the HRI shall appear on the label even if this results in no space for the AIDC. For other specific requirements related to the UDI carrier, please consult Section 4 of Annex VI Part C of the two Regulations.
For single-use devices of classes I and IIa medical devices and class A and class B IVD medical devices packaged and labelled individually, the UDI carrier shall not be required to appear on the packaging, but it shall appear on a higher level of packaging, e.g. a carton containing several (individually packaged) devices. However, when the healthcare provider is not expected to have access, in cases such as in-home healthcare settings, to the higher level of device packaging, the UDI shall be placed on the packaging (of the individual device).
For devices exclusively intended for retail point of sale, the UDIPIs in AIDC shall not be required to appear on the point of sale packaging. If the UDI carrier is readily readable or, in the case of AIDC, scannable, through the device’s packaging, the placing of the UDI carrier on the packaging shall not be required.
What changes in the medical device would require a new UDI-DI?
A new UDI-DI shall be required whenever a change could lead to misidentification of the device and ambiguity in its traceability. In particular, a new UDI-DI shall be necessary in the case of any change of the following elements: name or trade name, device version or model, labelled as single-use, packaged sterile, need for sterilisation before use, the number of devices provided in a package, critical warnings or contra-indications and CMR/Endocrine disruptors. A UDI-DI shall be associated with one and only one Basic UDI-DI.
Is the software subject to UDI rules?
The UDI shall be assigned at the system level of the software. The only commercially available software and software that constitutes a device in itself shall be subject to that requirement. The software identification shall be considered the manufacturing control mechanism and shall be displayed in the UDI-PI. UDI requirements for software are laid down in Annex VI Part C of the two medical device Regulations. A dedicated guideline with additional information on this aspect is available in MDCG 2018-5 guidance document.
What is the mandatory deadline for a device to comply with the UDI requirements?
The obligation for UDI assignment applies from the date of application of the two new Regulations, i.e. 26 May 2021 for medical devices and 26 May 2022 for In Vitro diagnostic medical devices.
The obligation for submission of UDI data in the EUDAMED database applies from 26 November 2022 for medical devices and 26 November 2023 for in vitro diagnostic medical devices (provided that EUDAMED is fully functional before the date of application of the respective Regulation; otherwise, this obligation applies 24 months after EUDAMED has become fully functional)
However, manufacturers will be able to voluntarily comply with registration obligations from 26 May 2021 for medical devices and 26 May 2022 for In Vitro diagnostic medical devices. It shall be noted that, provided that Eudamed is fully functional, at any time after 26 May 2021 for medical devices and 26 May 2022 for In Vitro diagnostic medical devices, the complete registration of devices (Article 29 of MDR and Article 26 of IVDR) remains a pre-condition for the possible registration of their relevant serious incident in Eudamed.
The obligation for placing the UDI carrier applies according to the following timelines:
The device as per Regulation (EU) 2017/745 (MDR)
Implantable devices and Class III devices
Class IIa and Class IIb devices
Class I devices
Placing UDI-carriers on the labels of devices MDR Article 123(3)(f), Article 27(4)
26 May 2021
26 May 2023
26 May 2025
Direct marking of the reusable devices MDR Article 123(3)(g), Article 27(4)
26 May 2023
26 May 2025
26 May 2027
The device as per Regulation (EU) 2017/746 (IVDR)
Class D IVDs
Class C and B IVDs
Class A IVDs
Placing UDI-carriers on the labels of devices IVDR Article 113(3)(e), Article 24(4)