

The IVD medical devices Regulation (EU) 2017/746 (IVDR) brings EU legislation into line with technical advances, changes in medical science, and progress in law-making.
The new Regulation creates a robust, transparent, and sustainable regulatory framework recognised internationally, improving clinical safety and creating fair market access for manufacturers.
In contrast to the previous Directive, the new Regulation is directly applicable and does not need to be transposed into national law. Therefore, the IVDR will reduce the risks of discrepancies in interpretation across the EU market.
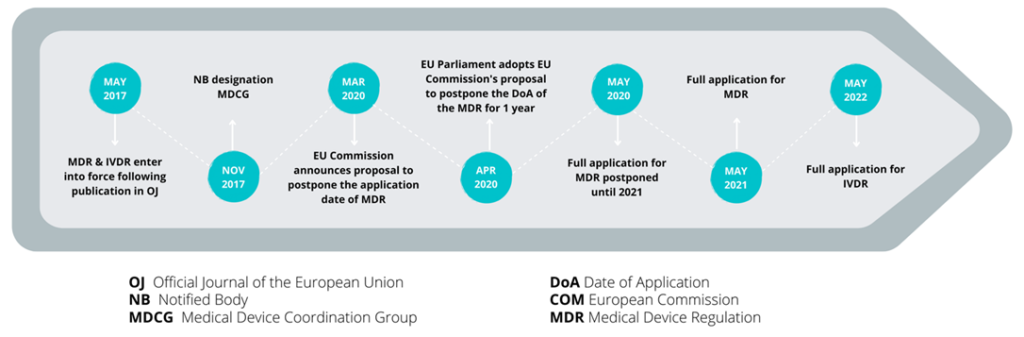
The IVDR will replace the existing in vitro diagnostic medical devices Directive (98/79/EC) (IVDD) and was published on 25th May 2017, marking the start of five years of transition from the IVDD, which is due on 25th May 2022.
Ready to Streamline Your Regulatory Compliance?
Join hundreds of companies who trust OMC Medical for their regulatory needs. Get expert guidance and ensure compliance across all markets.
Call Now +44 208 066 7260Compared to the current Directives, the new Regulation emphasises a life-cycle approach to safety, backed up by clinical data. The Regulation adds more stringent rules for the designation of Notified Bodies.
For national competent authorities and the Commission, they add more control and monitoring requirements. The Regulations clarify the obligations of manufacturers, authorised representatives, importers, and distributors.
Risk classification based conformity assessment
For IVDs, the most significant change concerns the risk classification of in vitro diagnostic devices and the role of Notified Bodies. As a result, around 85% of all IVDs will need oversight from Notified Bodies, compared to 20% under the Directive.
The IVDR also tightens the requirements for clinical evidence and conformity assessment.
For IVDs, most Class A devices can be self-certified by their manufacturers unless they are sold sterile. Devices in Classes B, C and D will require a conformity assessment by a Notified Body.
The conformity assessment of Class D devices will require the involvement of an EU Reference Laboratory designated for that type of device to verify the performance claimed by the manufacturer and compliance with the applicable Common Specifications.
For innovative Class D devices where no Common Specifications exist, an independent expert panel must provide its views on the performance evaluation report of the manufacturer.
General obligations of manufacturers
- Manufacturers must ensure that their products have been created and manufactured before releasing them to the market or putting them into operation by the requirements of this Regulation.
- Manufacturers must ensure that their products have been created and manufactured before releasing them to the market or putting them into operation.
- Manufacturers shall conduct a performance evaluation, including a PMPF.
- Manufacturers shall draw up and keep the technical documentation for those devices up to date. The technical documentation should allow the conformity of the device with the requirements of this Regulation to be assessed.
- Manufacturers of devices other than performance study devices must prepare an EU declaration of conformity and affix the CE marking of conformity when compliance with the applicable requirements has been established through the appropriate conformity assessment method.
- Manufacturers must adhere to the UDI system’s requirements as well as the registration requirements.
- The technical documentation, the EU declaration of conformity, and, if appropriate, a copy of the relevant certificate, including any revisions and supplements, must all be kept by the manufacturer. It shall be available to competent authorities for at least ten years after the final device covered by the EU declaration of conformity is put on the market.
- Manufacturers must have procedures in place to guarantee that series manufacturing meets the criteria of this Regulation. Any changes made to the product shall be considered promptly.
The traceability of devices using a Unique Device Identification system (UDI system) based on international guidance should significantly enhance the effectiveness of post-market safety-related activities for devices, owing to improved incident reporting, targeted field safety corrective actions, and better monitoring by competent authorities.
It should also help reduce medical errors and fight against falsified devices. One key aspect in fulfilling the objectives of this Regulation is the creation of a European database on medical devices (EUDAMED).
The EUDAMED should integrate different electronic systems to collate and process information regarding devices on the market and the relevant economic operators, certain aspects of conformity assessment, notified bodies, certificates, performance studies, vigilance and market surveillance.
The proper functioning of notified bodies is crucial for ensuring a high level of health and safety protection and citizens’ confidence in the system.
Designation and monitoring of notified bodies by the Member States, per detailed and strict criteria, should be subject to controls at the Union level.
FAQs (Frequently Asked Questions)
What is the timeline and transition to the new IVD Regulation?
Why are the key changes brought into the IVD Regulation?
- Classification system – the IVDR introduces a rule-based classification system for IVDs. IVDs will now be classified into four different classes based on risk, from class A (low) to class D (high).
- Changes to conformity assessment procedures – IVDs will now be subjected to conformity assessment based on the classification of the device. Classes B, C, and D IVDs will all require assessment and certification by a notified body for medical devices (appropriately designated for IVDs) prior to being placed on the market. This represents a significant change in today’s system, where many IVDs are self-declared devices rather than being assessed by a notified body.
- Performance evaluation and clinical data requirements – the requirements for performance evaluation of IVDs are defined in much greater detail in the new Regulations. Specific requirements are also defined in relation to the use of clinical data for IVDs and the conduct of clinical performance studies.
- Changes to requirements for in-house manufacturing of IVDs – under the existing legislation, IVDs which are manufactured within a healthcare institution and for use within that health institution are exempted from the Directive. Such tests may be developed due to the lack of a commercially available alternative, e.g., for rare diseases. The new Regulation places requirements on ‘in-house’ IVDs and the healthcare institutions which manufacture them and allows the introduction of additional requirements at the national level by individual member states.
Which obligations of the Regulations do manufacturers need to fulfil in order to place compliant devices on the market before the DoA?
Manufacturers should make sure to meet most obligations as possible, bearing in mind that the complete MDR/IVDR infrastructure, including EUDAMED, may not be fully functional before the respective DoA.
The Regulation applies to both the gadget and the maker. Manufacturers should do a compliance assessment of their products.
Does the role of Authorised Representative for IVDR differ from that of a Medical Device?
The role of Authorised Representative for IVDR and medical devices are the same:
- The manufacturer of a device is not established in a Member State. The device may only be placed on the Union market if the manufacturer designates a sole authorised representative.
- The designation shall constitute the authorised representative’s mandate; it shall be valid only when accepted in writing by the authorised representative and effective at least for all devices of the same generic device group.
- The authorised representative shall perform the tasks specified in the mandate agreed between it and the manufacturer. Upon request, the authorised representative shall provide a copy of the mandate to the competent authority.
- The mandate referred to in paragraph 3 of this Article shall not delegate the manufacturer’s obligations laid down in Article 10(1), (2), (3), (4), (5), (6), (8), (9), (10) and (11).
- Without prejudice to paragraph 4 of this Article, where the manufacturer is not established in a Member State and has not complied with the obligations laid down in Article 10, the authorised representative shall be legally liable for defective devices on the same basis as, and jointly and severally with, the manufacturer.
- An authorised representative who terminates its mandate on the grounds referred to in point (h) of paragraph 3 shall immediately inform the competent authority of the Member State in which it is established and, where applicable, the notified body that was involved in the conformity assessment for the device of the termination of the mandate and the reasons therefor.
- Any reference in this Regulation to the competent authority of the Member State in which the manufacturer has its registered place of business shall be understood as a reference to the competent authority of the Member State in which the authorised representative, designated by a manufacturer referred to in paragraph 1, has its registered place of business.





