

On 5 May 2017, the EU published the new EU MDR 2017/745 and IVDR 2017/746 regulations in which they formally introduced the UDI system in the EU. The UDI comprises the following components
- A device identifier (UDI-DI)
- A production identifier (UDI-PI)
The Basic UDI-DI is a technique introduced by the EU for linking medical devices to their regulatory documentation so that the model of the product can be uniquely identified throughout its entire lifecycle.
The linked documentation may include the declaration of conformity, notified body certificates, technical documentation and a summary of the safety and clinical performance of the device.
The UDI-DI identifies specific, detailed information about a particular device. The UDI-DI is a unique numeric or alphanumeric code specific to a device model and used as the ‘access key’ to the information stored in a UDI database.
Ready to Streamline Your Regulatory Compliance?
Join hundreds of companies who trust OMC Medical for their regulatory needs. Get expert guidance and ensure compliance across all markets.
Call Now +44 208 066 7260If any of the below details change, the device will then need a new UDI-DI.
- Name or trade name of the device
- Device version or model
- Labelled as a single-use device
- Packaged as sterile
- Maximum number of uses
- Need for sterilization before use
- Quantity of devices provided in a package
- Critical warnings or contra-indication
- CMR/endocrine disruptors
Before placing a device for clinical or performance evaluation on the market, the manufacturer has to assign a Basic UDI-DI to the device and enter it into the UDI and Device Registration module of EUDAMED, along with other data elements related to that device.
The Basic UDI -DI should be maintained by the manufacturers by establishing processes for the assignment and maintenance of Basic UDI-DIs.
According to the new rules, any manufacturer can assign a UDI to the device and to all the higher levels of packaging of the device as well before the device is placed on the market. The label of the device should have the UDI carrier.
The manufacturer has to ensure that before the device is placed on the market, the information related to the device has to be submitted correctly and saved to the UDI database. In the EU, the manufacturer has to assign a UDI along with a Basic UDI-DI to all his devices.
Some important points on the UDI:
- The UDI-DI have to be unique at each level of device packaging.
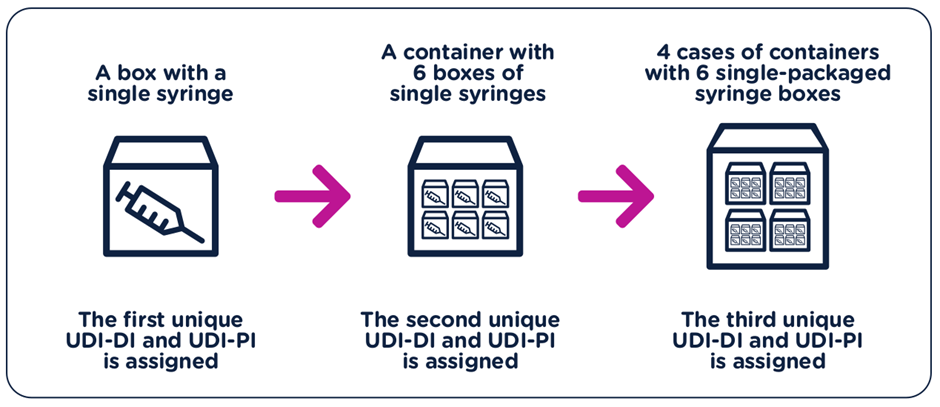
- Each component considered to be a device and is also commercially available on its own must be assigned a separate UDI unless the components are part of a configurable device that is marked with its own UDI.
- A new UDI-DI is required whenever there is a change that could lead to misidentification of the device or poses an error for its traceability.
- The data for new UDI-DIs shall be available at the time the device is placed on the market.
- Manufacturers shall update the relevant UDI database record within 30 days of a change being made to an element, which does not require a new UDI-DI.
Article 27 of 2017/745 and Article 24 of Regulation 746/2017 lay down that the UDI system shall consist
- Of production of a UDI that comprises a UDI device identifier (‘UDI-DI’) specific to a manufacturer and a device, providing access to the information and a UDI production identifier (‘UDI-PI’) that identifies the unit of device production and, if applicable the packaged devices
- Placing the UDI on the label of the device or its packaging
- Storage of the UDI by economic operators, health institutions and healthcare professionals, in accordance with the conditions laid down in the article.
FAQ
What is the UDI?
The UDI is a series of numeric or alphanumeric characters created through a globally accepted device identification and coding standard.
It allows the unambiguous identification of a specific medical device on the market. The UDI is comprised of the UDI-DI and UDI-PI. The unique identifier may include information on the lot or serial number and be able to be applied anywhere in the world.
The production of a UDI comprises the following:
- A UDI device identifier (‘UDI-DI’) specific to a device, providing access to the information laid down in Part B of Annex VI.
- A UDI production identifier (‘UDI-PI’) identifies the unit of device production and, if applicable, the packaged devices, as specified in Part C of Annex VI.
What is the Basic UDI-DI?
The Basic UDI-DI is the primary access key for device-related information in the Eudamed database, and it is referenced in relevant documentation [e.g. certificates (including certificates of free sale), EU declarations of conformity, technical documentation, and summaries of safety and (clinical) performance]).
- Its goal is to find and connect devices that have the same intended purpose, risk class, and key design and manufacturing features.
- It is distinct from the device’s packaging/labelling and does not appear on any commercial item.
- Any Basic UDI-DI must uniquely identify the devices (group) covered by the Basic UDI-DI.
What are the entities that provide the service of issuing UDI to medical devices?
- GS1
- Health Industry Business Communications Council (HIBCC)
- ICCBBA
- Informationsstelle für Arzneispezialitäten — IFA GmbH




How to get a Basic UDI-DI?
Basic UDI-DI should be assigned in compliance with the rules of the chosen issuing entity.
Can one Basic UDI-DI be linked with more than one UDI-DI?
Medical devices with the same intended purpose, risk class, essential design etc., are grouped under a single and unique Basic UDI-DI. As such, a medical device that has been assigned a UDI-DI can be linked to only one Basic UDI-DI. There is, therefore, a one-to-one relationship of 1 UDI-DI per 1 Basic UDI-DI. On the other hand, one Basic UDI-DI can be linked to multiple UDI-DIs. Therefore, there is a relationship of 1 Basic UDI-DI to N UDI-DIs.
Which products are subject to the UDI system?
The UDI system should apply to all devices, except custom-made and performance study/investigational devices.
Who is responsible for placing the UDI carrier on the device itself, on the label and the device’s package?
All UDI-related requirements are the responsibility of the manufacturer. This comprises assigning the UDI (and Basic UDI-DI), registering the UDI (and Basic UDI-DI) in the Eudamed database, and placing the UDI carrier on the device’s label, packaging, or, in the case of reusable devices, on the device itself.
What are the obligations of economic operators and health institutions in relation to UDI?
According to the two medical devices Regulations, manufacturers shall be responsible for the UDI assignment and placement of the UDI carrier, the initial submission and updates of the identifying information and other device data elements in the Eudamed database.
Manufacturers shall update the relevant database record within 30 days of a change being made to an element which does not require a new UDI-DI.
Distributors and importers shall verify that, where applicable, a UDI has been assigned by the manufacturer. All economic operators and health institutions shall store and keep preferably by electronic means the UDI of the devices, which they have supplied or with which they have been supplied if those devices belong to class III implantable devices.
Please note that the Commission may decide to adopt implementing acts to expand the scope of devices for which economic operators shall store and keep the UDI.
What is the procedure for systems and procedure packs to undergo a UDI registration?
Systems and procedure packs shall undergo a UDI registration, as described in MDR Article 29(2).
Before placing on the market a system or procedure pack pursuant to Article 22(1) and (3), that is not a custom-made device, the system or procedure pack producer shall assign to the system or procedure pack in compliance with the rules of the issuing entity, a Basic UDI-DI and shall provide it to the Eudamed database together with the other relevant core data elements listed in the MDCG 2018-4 guidance document.
Are devices, which are compliant with the Medical Device Directives (MDD and AIMDD) and placed on the market after the application date of the Regulations (legacy devices), continue to be subject to UDI requirements?
In order to facilitate the transition to the new system, the new regulations allow manufacturers to place products on the market after the general application dates of the new Regulations (and until 26 May 2024 at the latest) under valid Directive certificates.
These legacy devices are not subject to UDI obligations, but they should be registered in the Eudamed database.
Timelines for registration as described under question 6 also apply to these products. More information on the operational aspects of the registration of legacy devices is available in the MDCG 2019-5 guidance document.
How should a UDI appear on the label or package of a device?
The UDI Carrier [Automated Identification for Data Capture (AIDC) and human-readable interpretation (HRI) representation of the UDI] shall be on the label or on the device itself and on all higher levels of device packaging.
In the event of significant space constraints on the unit of use packaging, the UDI carrier may be placed on the next higher packaging level. Higher levels of packaging shall have their own unique UDI. Please note that shipping containers shall be exempted from the requirement.
The UDI must appear in a plain-text version/human-readable information (HRI) and in a form that uses AIDC technology. AIDC means any technology that conveys the unique device identifier or the device identifier of a device in a form that can be entered into an electronic patient record or another computer system via an automated process. The HRI consists of legible characters that people can easily read. If significant constraints limit the use of both AIDC and HRI on the label, only the AIDC format shall be required to appear on the label.
For devices intended to be used outside healthcare facilities, such as devices for home care, the HRI shall appear on the label even if this results in no space for the AIDC. For other specific requirements related to the UDI carrier, please consult Section 4 of Annex VI Part C of the two Regulations.
For single-use devices of classes I and IIa medical devices and class A and class B IVD medical devices packaged and labelled individually, the UDI carrier shall not be required to appear on the packaging, but it shall appear on a higher level of packaging, e.g. a carton containing several (individually packaged) devices. However, when the healthcare provider is not expected to have access, in cases such as in-home healthcare settings, to the higher level of device packaging, the UDI shall be placed on the packaging (of the individual device).
For devices exclusively intended for retail point of sale, the UDIPIs in AIDC shall not be required to appear on the point of sale packaging. If the UDI carrier is readily readable or, in the case of AIDC, scannable, through the device’s packaging, the placing of the UDI carrier on the packaging shall not be required.
What changes in the medical device would require a new UDI-DI?
A new UDI-DI shall be required whenever a change could lead to misidentification of the device and ambiguity in its traceability. In particular, a new UDI-DI shall be necessary in the case of any change of the following elements: name or trade name, device version or model, labelled as single-use, packaged sterile, need for sterilisation before use, the number of devices provided in a package, critical warnings or contra-indications and CMR/Endocrine disruptors. A UDI-DI shall be associated with one and only one Basic UDI-DI.
Is the software subject to UDI rules?
The UDI shall be assigned at the system level of the software. The only commercially available software and software that constitutes a device in itself shall be subject to that requirement. The software identification shall be considered the manufacturing control mechanism and shall be displayed in the UDI-PI. UDI requirements for software are laid down in Annex VI Part C of the two medical device Regulations. A dedicated guideline with additional information on this aspect is available in MDCG 2018-5 guidance document.
What is the mandatory deadline for a device to comply with the UDI requirements?
The obligation for UDI assignment applies from the date of application of the two new Regulations, i.e. 26 May 2021 for medical devices and 26 May 2022 for In Vitro diagnostic medical devices.
The obligation for submission of UDI data in the EUDAMED database applies from 26 November 2022 for medical devices and 26 November 2023 for in vitro diagnostic medical devices (provided that EUDAMED is fully functional before the date of application of the respective Regulation; otherwise, this obligation applies 24 months after EUDAMED has become fully functional)
However, manufacturers will be able to voluntarily comply with registration obligations from 26 May 2021 for medical devices and 26 May 2022 for In Vitro diagnostic medical devices. It shall be noted that, provided that Eudamed is fully functional, at any time after 26 May 2021 for medical devices and 26 May 2022 for In Vitro diagnostic medical devices, the complete registration of devices (Article 29 of MDR and Article 26 of IVDR) remains a pre-condition for the possible registration of their relevant serious incident in Eudamed.
The obligation for placing the UDI carrier applies according to the following timelines:
| The device as per Regulation (EU) 2017/745 (MDR) | Implantable devices and Class III devices | Class IIa and Class IIb devices | Class I devices |
| Placing UDI-carriers on the labels of devices MDR Article 123(3)(f), Article 27(4) | 26 May 2021 | 26 May 2023 | 26 May 2025 |
| Direct marking of the reusable devices MDR Article 123(3)(g), Article 27(4) | 26 May 2023 | 26 May 2025 | 26 May 2027 |





