Medical devices conforming with the medical device regulations must have a conformity marking. Medical devices that do not conform may still be placed in the market, provided they apply through the exceptional use devices pathway.
This article discusses the exceptional use requirements to be satisfied to gain access to EU and UK markets.
In the UK, the UKCA marking displays that the device conforms to UK Medical Device Regulations 2002. Without UKCA marking, the devices can only be placed in the market under certain circumstances.
Similarly, CE marking is accepted throughout the EU, which denotes that medical device conforms to the EU MDR 2017/745. This procedure may also apply to custom-made devices that have not complied with standard conformity procedures.
These devices are called special-purpose devices in the EU and are mentioned in Article 21 of EU MDR 2017/745.
The application form must be submitted for each different patient. MHRA approves the case-by-case application basis and may request more information if the application details are deemed unclear.
The application form for exceptional use devices requires the following details:
Risk analysis, hazard identification and estimation of risks
Name and address of the consultant
Patient’s medical condition details
Device necessity and
Other relevant information
Devices for special purposes in the EU
Devices under this category do not require a CE mark. Still, they must meet the conditions mentioned in EU MDR that the device is evaluated for its safety and effectiveness by the manufacturer.
Articles 62-82 of EU MDR describe the requirements to be met by manufacturers of investigational devices.
Firstly, for investigational devices, a proper clinical investigation plan is required. Read our detailed article on Clinical investigation in the EU for more information regarding this topic.
In short, the clinical investigation has a different technical file and application form than the CE-marked medical devices.
The application form should be submitted with the following documents.
Clinical investigation plan
Investigator’s brochure
Name and address of the manufacturer
Patient information sheet
Detailed risk analysis and test reports
Informed consent document
Other available technical documentation
Custom-made devices
As per EU MDR, ‘custom-made devices’ means as any device specifically made in accordance with a written prescription of any person authorised by national law by virtue of that person’s professional qualifications which gives, under that person’s responsibility, specific design characteristics, and is intended for the sole use of a particular patient exclusively to meet their conditions and needs.
However, the definition does not include ‘mass produced’ devices that need to be adjusted per user requirements. UK MDR 2002 5 (1) has a similar definition for custom-made medical devices.
The provisions in Great Britain (England, Wales and Scotland) can be accessed in Guidance on Custom-made devices in GB. Medical devices like dental appliances and prosthetics all come under custom-made devices as these are designed specifically for an individual and are not mass-produced.
Custom-made devices require technical documentation following Annex II and III of EU MDR. In addition to the documents, the manufacturer or authorised representative should draw a statement containing crucial information like that the device is intended for exclusive use by a particular user and data allowing unique identification of the medical device.
An important point to remember is that custom-made devices must conform to general safety and performance requirements in Annex I of EU MDR and essential requirements in Annex I Schedule 2A of the UK MDR 2002, and the devices are not exempt from these requirements.
FAQs
What devices come under custom-made devices?
Medical devices that come under Custom-made devices are specifically designed for an individual patient. Some great examples include dental crowns, fillings, elastics and retainers for dental structure or health. Other devices such as artificial eyes and maxillofacial implants like craniofacial implants, auricular, nasal implants, hearing aids and orthopaedic footwear fall into this category. The healthcare professionals must take accurate impressions of patients so that the device is a custom fit for the end-user or patient.
What is the difference between a custom-made device and an investigational device?
Investigational devices are those that are research devices that are developed that have no market equivalent. These devices are subjected to a clinical study to check product effectiveness. On the other hand, custom-made devices are not subject to clinical research but are customised according to the patient’s requirements. Both investigational devices and custom-made devices do not require CE/UKCA marking and require exceptional devices regulatory approvals to enter the respective markets.
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.
A Label is the written, printed, or graphic information that goes on the packaging of the medical device.
Instructions For Use (IFUs) or Package Insert is the essential information accompanying the medical device for its safe and effective use by the user. It can be a single to multiple-page document.
Labelling is the content that goes on the Label or IFUs.
What are the minimum requirements for labeling?
The ISO has published many standards applicable to the medical device industry. Some of them are as below:
Standard Number
Standard Name
ISO 18113
In vitro diagnostic medical devices – Information supplied by the manufacturer (labelling) – Part 1, 2, 3, 4 and 5
ISO 28219
Packaging – Labelling and direct product marking with linear bar code and two-dimensional symbols
Medical devices – Information to be supplied by the manufacturer
ISO 14025
Environmental labels and declarations – Type III environmental declarations – Principles and procedures
ISO 14021
Environmental labels and declarations – Self-declared environmental claims (Type II environmental labelling)
ISO 14020
Environmental labels and declarations – General principles
ISO 22742
Packaging – Linear barcode and two-dimensional symbols for product packaging
There are more specific product-oriented labelling standards available.
ISO 20417 has defines information to be disclosed by the manufacturer. Every medical device manufacturer, distributor, importer, or Authorized Representative is bound to comply with the standard before placing the device on market. The requirements are as follows:
Information on Label
Manufacturer details – Trade Name, address, country
Product description.
Product identification – model or catalogue number, Lot number, serial number, expiry date, UDI,
Storage instructions
Operating instructions
Warning or precautions
Presence of any harmful substances (>0.1% w/w), biological origin substances, medicinal substances, nanotechnology materials
Electronic IFUs (if available)
Mention of: Single-use/ Single patient multiple-use / Reuse / Limitation on reuse
If Sterile and method of sterilization
Explanation of safety-related colours
Information on Packaging
Name and address of the manufacturer or an authorized representative
UDI
Production controls – lot number, serial number, expiry date
Model number, catalog number, commercial name
Mention of: Single-use/ Single patient multiple-use / Reuse / Limitation on reuse
Storage or special handling requirements
Any special requirements for battery-powered medical device
Contraindications, warnings, or precautions
Information in IFUs
General information (as above)
Intended Use of the medical device
Safety information
Performance of the medical device
Any residual risk associated with the use of the medical device or its accessory
Any known contraindications
Document control number of the IFU
Safe disposal information
Any specific instructions for handling or preparatory treatment
Any warnings, precautions, or limitations
If any accessories or indicators are provided along with the device, instructions on their use to be provided in the IFU.
Technical description
The harmonized ISO standard makes sure true and uniform information is conveyed to a lay/common person.
Global Labelling Requirements
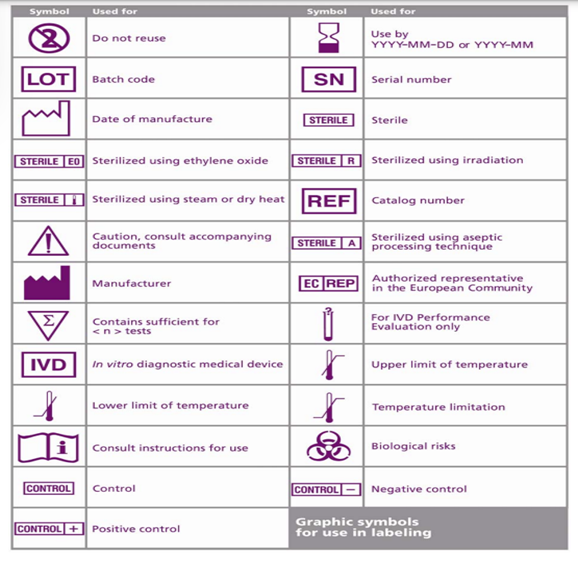
Most countries have a mandatory requirement for the IFUs or Labels in their local language. To streamline this requirement, ISO 15223 standard provides a list of signs and symbols that depict common terms such as Manufacturer, Lot number, storage conditions, Expiry, eIFU and many more.
The uniform symbols help in identifying the necessary information without the language barrier. Another advantage is it saves significant label space.
FAQs
Is it necessary to follow the ISO standards?
It is advisable to develop a medical device in compliance with the applicable harmonized standards. This shall favor in smooth marketing of the product along with its competitors.
Is it necessary to brief the symbols in IFU when symbols from standards are used?
Yes, it is required to brief every symbol in the IFU that is used on the label of the product.
Can a distributor or an importer label be affixed separately apart from the main label?
Yes, it is also allowed to affix these labels separately on the product. This is because one manufacturer may have several distributors or importers within EEA.
Is it necessary to create dedicated labels for accessories of medical devices?
Yes, it is. Not every time the accessory is shipped along with the medical device and it is required to identify them with appropriate labels.
If the manufacturer wants to provide an eIFU how to indicate this on the label?
Firstly, not all the medical devices are eligible for eIFU provision. Regulation 207/2012 states what are the categories of MDs that are eligible for eIFU.
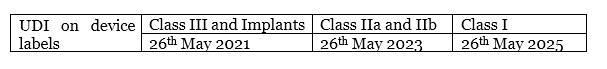
What is the deadline to implement UDI carrier on device labelling?
Article 123.3.f states these timelines as:
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.
With the UKCA deadline fast approaching and updating the existing regulation UK MDR 2002, MHRA has conducted a public opinion forum,
Some of the key highlights are stated below, for your quick review.
Transition Timeline
The significant update on this forum discussion is the Transition timeline of July 2023 has been moved.
The revised timeline is an extra 5 years for CE-marked products issued against EU MDR/IVDR, and an extra 3 years for CE marked under EU MDD and 5 years under EU IVDD.
There are a couple of caveats to this, but these are expected.
Devices that are subject to significant changes in design or intended purpose will be excluded from these provisions
All post-market requirements applicable to the new regulatory framework must be complied with for all products which benefit from the transitionary arrangements
MDSAP consideration for MHRA:
Additional to CE certification, UK MHRA will consider MDSAP certification as an alternative route to market to UK.
Caveat:
UK MHRA will require UK Approved bodies to consider MDSAP assessments.
Domestic assurance routers will allow an abridged assessment with scrutiny and Approved bodies will be able to reject if they consider sufficient evidence is not provided.
Classification:
Under the upcoming new regulation, MHRA is considering updating the Classification of Medical devices as well as In Vitro Devices(IVD) and Assisted reproductive technologies (ART)
And devices with nanoparticles will be moved between Class IIa -III depending on the risk involved.
Traceability:
MHRA will be bringing our requirements for economic operators to identify and record the following information:
Any economic operator to whom they have directly supplied a medical device
Any economic operator who has directly supplied them with a medical device
Any public or private sector health institution or healthcare professional to which they have directly supplied a medical
This will provide more traceability of the product.
Currently, this doesn’t state where and how the record needs to be maintained so we would ask the manufacturers to maintain this record and provide the same to us (UKRP) on a monthly basis for our records.
On the whole, MHRA is considering its new regulation to be in line with EU MDR/IVDR in certain aspects, we will know more when the new regulation is released and consider the above as MHRA’s view on changes to be made based on the public opinion.
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.
ISO – International Organization for Standardization, is the international, non-governmental body for drafting and establishing technical and non-technical standards.
These standards are developed by different committees within the International Organization for Standardization. Having around 165 member states, with one representative from each, International Organization for Standardization is a global entity catering to the needs of industry requirements.
Are ISO standards important?
The International Organization for Standardization medical device standards are the Bible for many countries, especially ones which do not have predefined regulations or processes.
In addition to general standards, ISO also publishes product-specific guidance such as for Implants, Orthopedic, Medical Electric Equipment, and many more.
Global International Organization for Standardization Requirements
In Europe, the European Commission has the Medical Device Regulation MDR 2017/745 and In-vitro Diagnostic Device Regulation IVDR 2017/746.
These regulations provide a detailed framework for introducing a medical device in the European market. However, in addition to that, certain International Organization for Standardization may also be referred to for ensuring a better-quality product.
Some of the many popularly used standards include:
ISO 14971:2019 Medical Devices – Application of Risk Management to medical devices
ISO 15223-1:2021 Medical devices – Symbols to be used with information to be supplied by the manufacturer – Part 1: General requirements
IEC 60601-2-83 Medical electrical equipment – Part 2-83: Particular requirements for the basic safety and essential performance of home light therapy equipment
IEC 60601-1 Medical electrical equipment – Part 1: General requirements for basic safety and essential performance
The European Commission also has Harmonized Standards, developed by European Standards Organization CEN, CENELEC, or ETSI, per the international standards.
It provides a list of the applicable harmonized standards for enhanced product safety and quality.
In the USA, the US Food and Drug Administration (FDA) has a Code of Federal Regulations (CFR) and Guidance.
CFRs are legally binding. Manufacturers must comply with the requirements of CFR
The guidance provides Agency’s thinking on regulatory issues. They are NOT legally binding
In addition to these, the FDA also accepts certain recognized consensus standards from different organizations such as International Organization for Standardization, CLSI, ANSI, IEC, CEN, etc.
These standards may be used to justify a Declaration of Conformity for a product. The widely accepted medical device International Organization for Standardization are, but are not limited to:
ISO 10993 – Biological Evaluation for Medical Devices
ISO 14160 – Sterilization of Healthcare Products
ISO 11737 – Sterilization of Medical Devices
In Canada, the Standards Council of Canada (SCC) is the International Organization for Standardization member body. Similar to the US FDA, the Therapeutic Products Directorate (TPD) of Health Canada periodically releases a list of acceptable international or national standards for medical devices.
Manufacturers can use these recognized standards in conjunction with the Health Canada’s Medical Devices Regulations (SOR-98/282) and the Guidance Documents, to prove product conformity and safe use in the market.
China‘s National Medical Products Administration (NMPA) is developing indigenous standards that more closely align with those of ISO. Biocompatibility testing is one avenue where the scope and requirements for China are more than that of the US/EU.
Hence, NMPA has developed various biocompatibility testing standards which are to be used in addition to the International Organization for Standardization standard.
For the rest of the world’s medical device industry,
India encourages International Organization for Standardization certification for all its industries. The medical sector must be International Organization for Standardization 13485 compliant while the pharmaceutical sector must be ISO 9001 compliant for Quality Management Systems, in addition to other relevant and applicable International Organization for Standardization.
Japan’s The Japanese Industrial Standards Committee (JISC) is an International Organization for Standardization member body. The regulatory authority, Pharmaceutical and Medical Device Agency (PMDA) revised its Ordinance No. 169 in 2021 to closely align with the International Organization for Standardization 13485:2016 standard. The transition period is 3 years and must comply by March 25, 2024
For the Korean regulatory authority, aligning the requirements of Korean Good Manufacturing Practice (GMP) to that of International Organization for Standardization 13485:2016 is believed to be a step closer to entering the Medical Device Single Audit Program (MDSAP)
Russia’s Federal Service for Surveillance in Healthcare (Roszdravnadzor) is known to accept International Organization for Standardization 13485:2016 certification. Information on acceptance of other International Organization for Standardization cannot be confirmed. It does not accept market approvals in the US, EU, or other countries as a reference for market authorization in Russia
Australia’s Standard Australia is a member of the International Organization for Standardization, IEC, and ICSID. It strongly encourages the use of international standards, except where their use is ineffective or inappropriate and does not develop any national Australian standard for which there is already an international standard in existence. In 2019, TGA published Therapeutic Goods (Conformity Assessment Standard for Quality Management Systems) Order 2019which provides a list of applicable conformity assessment standards.
Brazil’s ANVISA accepts Good Manufacturing Practices (GMP) along with the International Organization for Standardization 13485
FAQs
Can QMS be established solely based on ISO standards?
For countries that do not have their own QMS regulations, the ISO standard can be used as a reference. For countries with established local regulations, and that accepts ISO, both ISO standard and local/national regulations must be considered.
Are ISO standards freely available?
No. ISO standards are available for purchase from the ISO official website. However, they do have FREE read-only formats available.
Comparing ISO standards to local regulations, which one takes precedence?
The local or national regulation always takes precedence over the ISO standard.
Can the manufacturer use an older version of an ISO standard for compliance?
No. Manufacturers must make sure they comply with the active or most recent version of the ISO standard. This is not restricted just to ISO standards but applies to National regulations too. Manufacturers must keep their QMS up to date with the latest requirements of the industry. The ideal way to be updated is to refer to the latest version of any Standard or Regulation.
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.
Medical device nomenclatures are those products used to prevent, diagnose, treat, and monitor the many diseases known to humankind. Medical devices and medicines play an equally important role in treating human beings.
To learn more about medical devices, read our article on the definition of a medical device. This article discusses the nomenclature of medical devices and examples of these.
What is the Nomenclature of Medical Devices?
To simply put it, the nomenclature is the naming of a medical device. Although medical devices are classified into different risk classes, they should be named so that it is universally identified. Standardised nomenclature facilitates this easy identification.
A medical device nomenclatures is needed to simplify trade and tracking among the different regulatory authorities, Ministries of Health, and other organisations that regulate medical devices.
A standardised medical device nomenclatures aids in the following aspects:
Grouping and classification of medical devices.
Registration under different regulatory bodies or Ministries of Health
Streamlined procurement and distribution
Grouping of medical devices in various electronic health records and medical device databases
Vigilance reporting, field safety and post-market surveillance
The different medical device nomenclatures available are as follows:
GMDN or Global Medical Device Nomenclatures.
EMDN or European Medical Device Nomenclatures
UMDNS or Universal Medical Device Nomenclatures System
Other nationally developed nomenclature systems
Global Medical Device Nomenclatures (GMDN)
About 10% of countries use Global Medical Device Nomenclatures worldwide. It is a system of internationally accepted descriptors used to identify medical devices. The GMDN Agency manages GMDN codes, a non-profit organisation.
GMDN is a 5-digit code containing the following information:
GMDN Term Name: Anaesthesia ventilator
GMDN Code: 34851
GMDN Definition: A mains electricity (AC-powered) stand-alone, automatic cycling device used to assist and control alveolar ventilation during general anaesthesia and is compatible with inhaled anaesthetic agents. It has fewer functions and is less complex to operate than an intensive care ventilator but adequately meets the patient’s ventilation needs for oxygen (O2) and carbon dioxide (CO2) exchange to maintain normal blood gas concentrations. The device provides a mechanical means to deliver the breathing gas to the patient in a controlled pattern. It is equipped with alarms to warn of changes in respiration or the onset of unsafe operating conditions.
GMDN was introduced for a variety of regulatory purposes. GMDN is based on the ISO 15225: Medical device nomenclature data structure’. Read more about the frequently asked questions about GMDN here.
European Medical Device Nomenclature (EMDN)
European Medical Device Nomenclature or EMDN is introduced due to Article 26 of EU Regulation 2017/745 of Medical devices and Article 23 of EU Regulation 2017/746 of in-vitro diagnostic medical devices.
Like GMDN, it plays a considerable role in device nomenclature and serves various regulatory purposes. One of the primary uses is while registering a medical device in EUDAMEDwhere it is closely linked to UDI-DI.
Structure of EMDN
The European Medical Device Nomenclature is characterised by its alphanumeric structure and is established in a seven-level hierarchical tree where it clusters medical devices into three primary levels:
Categories: the first hierarchical level – alphanumeric.
Groups: the second hierarchical level – 2 numbers indicating group.
Types: the third hierarchical level – a series of numbers 1,2,3,4 and 5.
EMDN was adopted from the Classificazione Nazionale Dispositivi medici (CND) classification. The EMDN can be accessed at the EMDN list. European Medical Device Nomenclature categorises into three primary levels, categories, groups, and types.
A category comprises several groups composed of various kinds of medical devices.
Source: https://webgate.ec.europa.eu/dyna2/emdn/. In the above image of EMDN, ‘A’ is the category, ‘A01’ is Group and ‘A0101’ to ‘A0199’ are the types of medical devices.
Universal Medical Device Nomenclature System (UMDNS)
Universal Medical Device Nomenclature System or UMDNS was developed by the Emergency Care Research Institute (ECRI). Many nations have adopted this standard around the world. UMDNS is used in inventory control, work order control, and regulatory systems applications.
It is a 5-digit code unique code and a term for different types of medical devices. One can find the UMDNS code list here. UMDNS is updated monthly.
CND Nomenclature
CND nomenclature or ‘Classificazione Nazionale Dispositivi medici’ was developed by Italian Ministry of Health. In addition to Italy, it is also used in Portugal and Greece.
The guidance document on CND nomenclature explains the basic principles and structure of CND, which also applies to EMDN as EMDN was adopted from CND nomenclature. Following this, medical devices are clustered into three levels:
Category
Group
Type
FAQs
Is UDI the same as GMDN?
Both Unique Device Identification System (UDI) and GMDN are used in device identification. However, the two have some fundamental differences. UDI is inferior because of its lack of unity. It does not have a structure; therefore, device identification becomes more difficult with UDIs. Nonetheless, it is an effective tool for the traceability of medical devices. FDA utilises UDIs for medical device identification. The user guide to GUDID explains how the UDIs are managed in US FDA’s database, GUDID.
Can EMDN be accessed free of charge?
The EMDN is accessible to all stakeholders- free of charge. Hence, it can be utilised by a non-exhaustive list of stakeholders such as manufacturers, patients, research organisations, practitioners, hospitals, etc. The EMDN can also be downloaded from here.
Is there a guidance document that helps economic operators to map the EMDN information into the forthcoming EUDAMED database?
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.
The EU MDR 2017/745 imposes more stringent requirements for Class I devices. Under the new regulations, Manufacturers must righty classify a medical device and provide technical documentation following the device class.
The risk class under MDD could change under MDR. In some cases, medical devices could be up classified from Class I medical devices to Class II a/b medical devices or Class III medical devices.
MDCG guidance on Class I medical devices is a valuable resource document for Class I medical devices Manufacturers. Manufacturers can place the devices by following the steps mentioned below.
Steps for placing a Class I Medical Devices
Step 1: Integrate MDR into the existing Quality Management System.
This allows the correct documentation to be created following the risk classification of the device.
Step 2: Confirm whether the product is a medical device under the scope of MDR.
Confirm that the product meets the definition of a medical device as stated in Article 2 of EU MDR based on its intended use and primary mode of action. In some cases, the product may be out of the scope of MDR and may be termed as a medical device ‘accessory ’. EU MDR also states how accessories for a medical device is also to be classified on its own apart from the main medical device.
Step 3: Confirm whether the medical device is a Class I medical devices.
It must be noted that several MDD Class I medical devices will be reclassified under the MDR as per the new classification criteria in Annex VIII, such as most software (rule11) and Class I medical devices made up of chemicals or combinations of substances (rule 21).
Step 4: Pre-market procedures.
Verify that the device meets the General Safety and Performance Requirements (GSPR).
A proper and well-established risk management system should be implemented. Manufacturers must ensure that the associated risks are identified, analysed and appropriate corrective actions are taken.
This must be guaranteed throughout the product’s lifecycle and documented. When standard specifications are available, Manufacturers must adhere to them unless they can demonstrate that they have adopted a solution that is at least as safe and effective.
The Manufacturer should periodically update the clinical evaluation report, risk management and post-market reports.
Conduct Clinical evaluations
All devices require clinical evaluation, Class I medical devices are not exempted from this requirement. A clinical evaluation report must contain essential information such as device descriptions, literature reports and post-market surveillance reports to name a few.
MDR also emphasises the need to consider available alternatives and the acceptability of the benefit-risk ratio.
Prepare an up-to-date technical file.
The Manufacturer will draw up the technical documentation that demonstrates the conformity of their devices with the specific requirements of the MDR. Technical documentation is mentioned in Annex II and III of EU MDR.
The Manufacturer is obligated to keep the file updated and make them available to competent authorities, notified bodies and the authorised representative.
The technical file must contain the following elements. The detailed information can be found in Annex II of EU MDR.
Device description and specification, variants, and accessories
Information to be supplied by the Manufacturer
Information on Design and manufacturing
General Safety and Performance Requirements
Benefit-risk analysis
Risk management
Verification and validation of product
Demonstration of Conformity
The documents in the technical file should be made available in the language accepted in the Member State in which the device is sold. Please read our article on EU Requirements for translations to understand this requirement specified in EU MDR.
Involvement of Notified Bodies
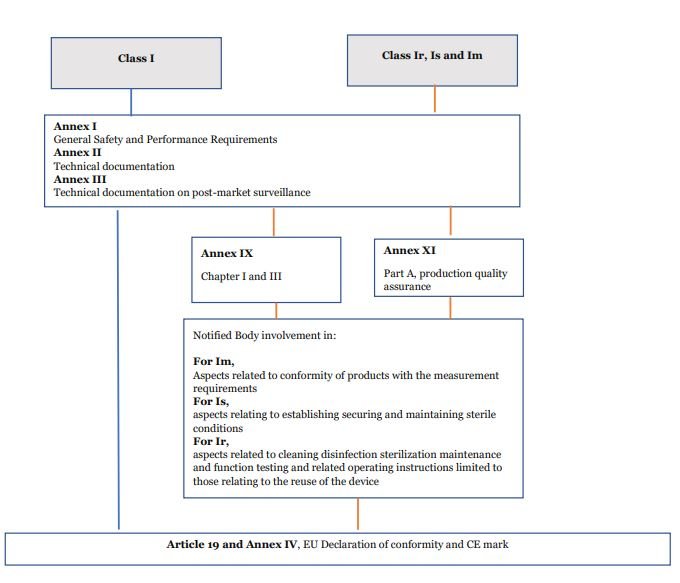
Most Class I devices do not require the involvement of a Notified Body. However, device Classes ‘Ir’, ‘Is’ and ‘Im’ have this requirement. ‘Ir’ devices are those which are reusable instruments. ‘Is’ are devices to be used in a sterile condition. ‘Im’ are those devices with a measuring function.
For these class I medical devices, the involvement of Notified Bodies is limited to audit and the NB assesses those specific controls. For example, an “Is” device assessment of NB would involve auditing the Sterilisation process and controls.
Prepare Instructions for Use and Labelling
Medical device manufacturers must ensure that the instructions for use and labels are available in languages accepted within the Member State they decide to market in.
The instruction for use must indicate the safety and performance information so that its users are aware of its intended use. The instructions for use (IFUs) should have drawings and internationally accepted symbols.
The Declaration of conformity (DoC) is a document signed by the Manufacturer that states the device fulfils the requirements set forth by MDR. For more information, read our article on Declaration of conformity.
Step 6: Affix CE mark.
CE mark should be placed on the medical device to show compliance. If it cannot be placed on the product itself, it must be placed on the packaging.
Before placing in the EU market, all devices must be registered on EUDAMED. EUDAMED is a secure, web-based European Databank on Medical Devices. Although Class I devices are not much of a threat to human life, they must be made available on EUDAMED. It ensures that the device information is accessible to the public.
Transitional provisions for Class I Medical Devices
Transitional provisions are in place to help manufacturers transition to MDR from the directives. Article 120 of EU MDR gives an idea of the exemptions from MDR and greatly helps manufacturers by providing more time to meet the requirements.
The following conditions must be met to be able to market a device according to Directive 93/42/EEC until 26 May 2024:
The device complies with Directive 93/42/EEC.
The device has not undergone significant changes in device design or intended purpose.
The device has a valid Declaration of Conformity (DoC)
The post-market surveillance, Market Surveillance, Vigilance and Registration of economic operators and devices requirements are met.
FAQs
Does a Class I Device manufacturer require an ISO certificate for the Quality Management System?
Not necessarily. The Class I manufacturer must have an established QMS, and obtaining a certification is not mandated. Although globally, there is a significant benefit for the Manufacturer to hold an ISO QMS certification. Major markets worldwide accept ISO 13485 certificate as one of the key entry factors into their geography. A few key markets, such as Canada, Japan, Australia, Singapore, and Malaysia, require ISO Certification. The ISO 13485 certificate is recognised worldwide as a significant standard for a quality management system for medical device manufacturers.
Should Class I Device DoC contain the reference to MDR Annex (s)?
Yes, but this has been considered of less importance while drafting the DoC. Declaration of Conformity (DoC) must mention the respective Annex(s) in the MDR that it has complied with for any device class. This is one of the required information to be entered while performing product registrations in many countries.
What is the conformity procedure for Class I devices?
The conformity procedure for all Class I devices is mentioned in the flow chart.