PMCF is a continuous process that updates the manufacturers’ clinical evaluation during the Post-market surveillance. They must collect user feedback, clinical data, and all other clinical experiences.
This article provides an overview of the design, implementation, and appropriate use of PMCF studies. these are an integral part of PMS reports.
There may be some data limitations during the premarket phase, like the duration of the investigation and the number of subjects involved or restrictions in the applicability of clinical data. Also, a complete diagnosis of all the potential risks and benefits is impossible during the premarket phase.
Post Market Clinical Follow-up (PMCF) is a study to answer specific questions like safety, clinical performance, or effectiveness of the medical devices according to their labelling from the routine use of the clinical practice.
PMCF studies address the uncertainties in the benefits or risks of medical devices like:
Unanswered questions about long-term safety, performance, and effectiveness of the clinical data
Novel/Innovative technologies used in the design, materials/products, and principles of the medical devices
Uncertainties in generating results from the study population to other populations like adults to children
Urgent market access due to emergencies like pandemics
Rare adverse events which can be identified using large datasets
Adequacy of mitigation
PMCF studies/reports must be conducted based on applicable laws, regulations, and ethical requirements. The personal information about the patient should be very confidential, and the documents must be approved with ethics committee under proper consent. Specific guidance and standards to be followed like:
Clearly stated objectives
Scientifically sound study design
Appropriate study plan
Design and Implementation
Factors that need to be considered during the design of PMCF studies:
The study settings such as locations must be clearly described
Study population like inclusion and exclusion criteria should be clearly described
Comparison/Control groups should be justified
The sample size should be clearly stated
Adverse events, risk factors, confounding factors and all other measures should be identified
To minimize the loss, frequent follow-up of the type and duration of the patient
Statistical analysis like missing data and modifications should be clearly described
Factors to be considered during the implementation of the PMCF study plan:
Data collection: Data should be evaluated according to the validated measurement methods
Quality control: Ensure good quality, proper training, selection, and supervision should be performed
Final report and interpretation: Demonstrating conclusion based on the original objectives or hypothesis
Based on all the above factors, there might be changes that impact the clinical evaluation and risk management process. Hence, the medical device must be reassessed to comply with the essential principles / general safety performance. Such assessments may include:
PMCF studies state that the data source collected from real-world clinical experience is essential. Examples of such data are as follows:
Patient-generated health data: Patient report outcome or health data collected from the patient, family members or their caregivers
Device registry: A system that collects data, results and the population that got exposed to that medical device
Health record: The medical record that the health care provider has maintained over the period
Administrative data: Data like health insurance claims
Survey data: Data collected by the survey from health professionals, patients/customers
Bias and Confounding
Potential bias is a deviation caused by overestimating or underestimating the treatment effects. Confounding is the distortion that occurs when the study group differs from the other factors. Some methods to control bias in PMCF study include:
Appropriate selection of study population
Use of validated survey instruments
Training the staff according to the standard
Avoid loss during the follow-ups
Selection of proper statistical data
FAQs
How is the PMCF study different from the clinical investigation?
PMCF, as it states, is the post-market study conducted for a CE-marked device to evaluate the device’s safety performance, whereas the clinical investigation is performed for a device to be CE-marked.
How much importance is given to a PMCF in the MDR?
PMCF is understood to be a continuous process in the device life cycle, and every class of device must have a PMCF plan established to proactively collect and evaluate the clinical data. The analysis of a study must be documented in the PMCF evaluation report, which is a part of the CER and the technical documentation.
What could go into a PMCF?
· Complete clinical data with user feedback · Any specific methods include during the PMCF study or survey · Clinical Evaluation Report (CER) · Address any harmonized standards used in the PMCF plan · PMCF objectives
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.
The MDR reinforces the clinical data and evaluation process (article 61 and Annex XIV), and the manufacturer must confirm the device’s conformity to fundamental health and safety requirements using reliable clinical data and evaluation.
The clinical evaluation establishes the device’s safety and capacity to fulfil its intended function. It also evaluates adverse side effects and determines whether the benefit-risk ratio is acceptable.
Manufacturers must plan, carry out, and document a clinical evaluation in line with Article 61 and Part A of Annex XIV.
Clinical data for the medical device are created, compiled, examined, and ultimately evaluated through a systematic and organised process called a clinical evaluation.
The Clinical Evaluation Report (CER), which the manufacturer uses to show that the medical device complies with the general safety and performance requirements specified in Annex I of the MDR, is the end result of the clinical evaluation.
The Clinical Evaluation Report (CER) is an essential component of a manufacturer’s quality management system and an essential component of the technical documentation for the medical device (MDR Article 10 (3)). It must be actively updated on a regular basis utilising information from the post-market clinical follow-up and post-market surveillance of the medical device (PMCF).
Thus, clinical evaluation is a continuous procedure throughout a medical device’s life cycle.
The Objective of Clinical Evaluation
The clinical evaluation aims to show that the medical device can be used as intended while still being safe and effective, including in terms of its clinical advantages.
The clinical evaluation can also be used to reevaluate risks and find previously overlooked hazards or dangers. The acceptability of hazards must be reevaluated by manufacturers using the most recent clinical evidence.
The objectives of the clinical examination include:
The product’s use for its intended purpose under normal circumstances demonstrates conformity with the general safety and performance requirements listed in Annex I of the MDR
Evaluating or excluding undesirable side effects
Proof of the validity of the risk-benefit ratio
Proving the makers’ medical claims.
Alternative product methods and technologies that can be used in place of the treatment being evaluated are evaluated and documented as part of the clinical study.
The clinical evaluation must ensure that the tested product is not worse than the potential substitutes. The clinical evaluation needs to describe and assess the state of the art.
When assessing state of the art, clinical benefits, safety, and performance should be taken into account. When designing and producing their products, medical device makers must take the latest technological advancements into account.
Clinical Evaluation Data
Clinical information gathered while using the medical device forms the basis of the clinical evaluation. The following are some potential sources for these:
Clinical trial(s) conducted by the manufacturer of the medical device
Clinical trial(s) or other research on a known similar product from the scientific literature
Data from post-market surveillance (PMS) are clinically significant, particularly from post-market clinical follow-up (PMCF).
Reports regarding additional clinical trials using the product under review or a comparable product that has been published in the peer-reviewed scientific literature
Manufacturers must consider preclinical data in addition to clinical data when making their clinical evaluations.
For instance, this comprises the outcomes of the following tests: Testing for biocompatibility, electrical and mechanical safety electromagnetic compatibility in accordance with IEC 60601-1-2, usability, software, animal, simulation, and laboratory testing, as well as testing for durability and stability.
For absolutely non-critical products (stand-alone software, dental drills, oral spatulas, etc.) and must be justified by the manufacturer based on risk management, in accordance with MDR Article 61 “Clinical Evaluation” Section 10.
The manufacturer’s claims, the anticipated clinical performance, and the precise interactions the device has with the human body are all taken into account in this explanation.
According to Annex II of the MDR, the manufacturer in this situation must explain in the technical documentation why they believe it is appropriate to show compliance with the general safety and performance requirements based solely on the outcomes of non-clinical test methods, including performance evaluation, technical testing, and pre-clinical evaluation.
Clinical Evaluation Plan (CEP)
A medical device’s clinical evaluation is a continuous process for developing, collecting, analysing, and evaluating clinical data. It is systematic and well-planned.
Manufacturers are required to create and update a clinical evaluation plan in accordance with Article 61 (paragraph 12) and Annex XIV Part A “Clinical Evaluation” of the MDR (CEP).
Basic ideas like the goals and format of the clinical evaluation are already stated in this strategy. The manufacturer establishes the fundamental performance and safety standards that relevant clinical data in the CEP must back up.
With detailed clinical outcome metrics, it outlines the desired clinical advantages for the patient and specifies the intended purpose, intended target groups, and explicit indications and contraindications.
A new required component of the clinical evaluation plan is a clinical development plan (CDP) for organising pertinently planned clinical trials, including a post-market clinical follow-up plan (PMCF plan).
These adjustments give clinical findings more weight. The Clinical Development Plan (CDP) explains how the manufacturer will gather new or extra clinical data through clinical trials or observational studies to solve open “gap analysis” problems at the beginning of the development phase.
Human volunteers are used in clinical trials to assess the clinical effectiveness and safety of medical equipment.
Class III Devices and Implantable Devices
Clinical investigations must always be carried out in the case of implanted devices and class III devices, with the following exceptions:
The already marketed device has been altered by the same manufacturer, who has also shown that the altered device is equivalent to the marketed one.
The notified body has approved of this demonstration, and the clinical assessment of the marketed device is sufficient to show that the altered device complies with the necessary safety and performance requirements.
Additionally, there is no requirement for clinical testing for class III and implantable devices if a manufacturer can show that its product is functionally equivalent to a product that has already been marketed, provided that the notified body has approved the demonstration and the following requirements are met.
The two manufacturers have a contract in place that expressly grants the manufacturer of the second product full access to the technical documentation on a continuing basis, and the original device manufacturer is still in business.
Additionally, No obligation for clinical investigation for Class III and implantable devices:
If the devices have been legitimately marketed under previous directives, the clinical evaluation is supported by enough clinical data, and they adhere to Common specifications where they are available.
Annex XIV
A clinical evaluation must be planned, continually carried out, and documented by manufacturers in order to:
Create and maintain a clinical evaluation plan,
Utilising a systematic, scientific literature study, determine the clinical data that is available that is pertinent to the device and its intended use, as well as any gaps in the clinical evidence;
Evaluate each relevant clinical study’s applicability for proving the device’s performance and safety;
To produce any additional or new clinical data required to address unresolved problems through adequately conducted clinical research in accordance with the clinical development strategy; and
In order to conclude the safety and clinical performance of the device, including its clinical advantages, all pertinent clinical data must be examined.
Equivalence
Equivalence for the EU MDR clinical evaluation must be proven in two distinct ways.
Clinical
Used for the same clinical condition (with equivalent severity and stage of disease).
Utilised for the same medicinal purpose, and utilised for the same intended purposes, and
Utilised at the same body location, and used in a population with similar features (e.g., age, gender, anatomy, physiology, etc.), and not anticipated to produce noticeably differing performances (in the relevant critical performances such as the expected clinical effect, the specific intended purpose, the duration of use, etc.).
Technical
Have similar specifications and properties (e.g., physicochemical properties such as type and intensity of energy, tensile strength, viscosity, surface characteristics, wavelength, surface texture, porosity, particle size, nanotechnology, specific mass, atomic inclusions such as nitrocarburising, oxidability),
Similar design
Used under the same conditions, similar deployment methods (if applicable), and similar operating principles).
Biological
Use the same tools or substances when in contact with the same body fluids or human tissues.
FAQs
What do you mean by clinical evaluation?
A clinical evaluation is a systematic and well-planned procedure used to acquire, gather, analyse, and ultimately evaluate clinical data for a medical device.
What is clinical evidence?
Clinical evidence is defined as clinical data and clinical evaluation results about a device of sufficient amount and quality to permit a qualified assessment of whether the device is secure and provides the expected clinical benefit(s) when used in accordance with the manufacturer’s instructions.
During the conformity assessment, the manufacturer must submit the complete information material (labelling, IFU (Instructions for Use), any promotional materials and other relevant documents), Clinical evaluation plan & report with the available clinical data, which includes (General Safety and Performance Requirements) GSPR, intended target groups and purpose, qualitative and quantitative aspects of clinical safety, performance procedures and other risk factors involved.
The manufacturer must carry out a gap analysis to fill the sufficient data according to the guidelines to meet the safety and performance of the device as in PART A Annex XIV in MDR 2017/745.
A clinical evaluation report (CER) is a technical document submitted by the manufacturer for the conformity of the devices. While updating the clinical evaluation, the manufacturer must consider,
MEDDEV Guidelines & Frequency of Updates
If the device carries any frequency risk during the intended use
If the device needs any changes like clinical sciences, materials, or other evaluation
Consider the data available on the PMCF (Post Market Clinical Follow-up) reports, clinical investigations, and other studies which may influence the frequency of updates.
Changes in the design or procedure
For the showing of equivalency – technological, biological, and clinical criteria must be considered.
Some general considerations during the Updates
PMS (Post Market Surveillance) always gets updated with new data like safety reports, published literature, registries, PMCF reports and other data during the intended purpose.
Those data should be fed into the clinical evaluation periodically. Also, check the benefit-risk ratios, undesirable side effects and other risk mitigations during the updates.
This may result in changes in the risk management reports, IFUs (Instructions for Use) and PMS activities as in PART B Annex XIV in MDR 2017/745.
Before conducting clinical evaluation and/or investigation for all class III and class IIb devices, the manufacturer is permitted to consult the expert panel mentioned in Article 106 MDR to review the manufacturer’s intended clinical development strategy and proposals for clinical investigation.
In particular, the criteria for an appropriate data set for assessment of the conformity of a device, regarding the clinical data necessary for clinical evaluation, physio-chemical characterization, and microbiological, biocompatibility, mechanical, and electrical considerations, shall be made available to the Member States, notified bodies, and manufacturers through the Commission.
The person who performs the clinical evaluation should know
Research Methodology
Information management
Regulatory requirements
Medical writing with a degree from higher education or 5 to 10 years of professional experience
The technology of that device and complete information
Diagnosis and management of medical alternatives with the standards
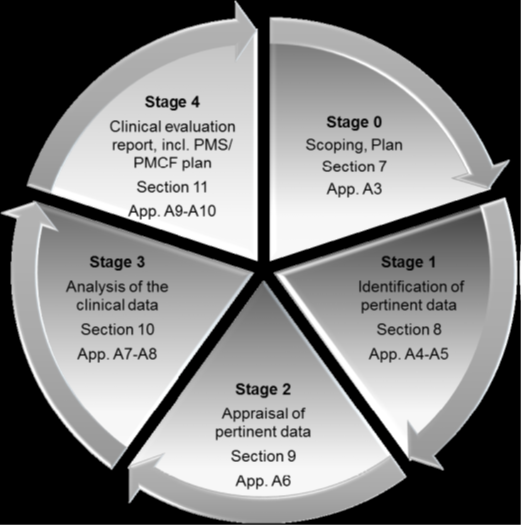
Source: MEDDEV 2.7 Clinical Evaluation (Stages of clinical evaluation)
Stage 0: Define the Scope
Basic and various aspects of the scopes are
Initial device description
Design features, intended purpose, applications of the device and other equivalence
Complete risk management documents which address clinical risks
Any changes in the design, material, or procedures. Also, any changes in IFUs, labels or other promotional materials
PMS aspects like new clinical data or knowledge which may change the state of the art and other aspects
It is crucial to acknowledge that there is a wide range of types, histories, and hazards associated with the technology employed in medical devices.
Since many devices have undergone incremental development or modification, they are not unique. It might be possible to use the clinical reports of an equivalent device’s performance and safety from experience and literature to establish the requirement for clinical data acquired by clinical trials by utilizing the clinical evidence investigation of the experimental device.
Stage 1: Identification of Pertinent data
Manufacturer-generated and stored data
All premarket clinical investigations, risk management activities and PMS programs like PMCF studies, vigilance reports, literature and compliance reports, field safety corrective actions, and other user reports.
Literature Information
Searching the literature reveals prospective sources of clinical data. The following aspects need to be considered.
Identify all the relevant favourable and unfavourable data
Obtain several types of searches like scientific literature databases, internet and non-published data and other literature that has a direct interest
Literature search and other appraisals of clinical data
The literature search must be thoroughly documented so that the methodology can be evaluated critically, and the findings can be confirmed, and the search can be repeated as needed.
Stage 2: Appraisal of Pertinent Data
The appraisal/evaluation plan includes
Criteria for judging the methodological quality and scientific validity of each data set
Standards for establishing the device’s intended use relevance to the clinical evaluation
Standards for weighing each data set’s contribution to the overall clinical assessment
The following section provides examples of factors that can be considered when assessing the methodological quality and scientific validity of the evidence, as in Article 61(3).
Pre- and post-market clinical investigation study plans like adequacy of sample size, random or blinding of patients, adequacy of the follow-up period, serious adverse effects reports and other medical interventions
Some additional aspects like a clinical investigation plan, case report form, audits, regulatory authority approvals, ongoing clinical investigation report which is conducted outside or in the territory and other gap analysis
Data obtained from case studies, patient dossiers, device registries, vigilance data, and other useful data
Data processing is like converting data to a standard format, and statistics
Good clinical practice with all the legal requirements
Sufficient description with proper disclosure of the report
When assessing the relevance of obtained data, we must consider whether the data are meant to directly demonstrate appropriate clinical performance and clinical safety of the device (commonly referred to as pivotal data) or whether they serve an indirect supportive role.
There is no one well-established approach for weighting clinical data due to the variety of medical devices. Instead, the evaluators should choose the best criteria to apply to each evaluation and adhere carefully to these pre-established criteria.
Stage 3: Analysis of Clinical Data
A device’s clinical efficacy and safety should not be demonstrated using data that are not methodologically sound (such as single patient reports).
The evaluators should employ reliable methodologies, conduct a thorough study, decide whether more clinical research or other actions are required, and identify PMCF needs to establish compliance.
During the evaluation, the evaluator should include
Pre-clinical testing
Benefits-risks associated with the patients
IFU correctly addresses risk mitigating methods
Gap analysis like identifying the entire range of products used during evaluation, conditions of use, the number of patients exposed, severity of adverse events, hazard analysis, current standard of care and other profiles
Some additional investigation into missing data and eradicating the issues of compliance.
The clinical evaluation report should have enough details for a third party to read and comprehend it (e.g., regulatory authority or notified body).
The manufacturer must demonstrate compliance with the general safety and performance requirements using clinical evidence, non-clinical data obtained from non-clinical testing methods, and other pertinent documentation.
These materials must also be included in the technical documentation for the relevant device.
Cross-references between the clinical evaluation report’s contents and the pertinent documentation supporting them are required. Which claims are supported by which pieces of data, and which express the evaluators’ opinions should be clear?
With cross-references to the location in the technical documentation from the manufacturer, the report should include references to literature-based data as well as the titles and investigational codes of any clinical investigation reports (if applicable and available).
FAQs
What are CEP and CER?
The clinical evaluation plan (CEP) outlines the devices that will focus on the CER, including their dimensions, intended uses, target population, and patient clinical benefits. Every time brand-new medical equipment is put on the market, the manufacturer must prove that it conforms to all applicable Essential Requirements (ERs) by using the proper conformity assessment methods.
What is the demonstration of equivalence?
When proving equivalence to another device, the MDR stipulates those technical, biological, and clinical features. Although MEDDEV 2.7 Appendix 1 describes these general qualities and aligns them with the MDR Annex XIV Part A (3) requirement, the requirements for each of the three characteristics differ.
What do you mean by similar devices?
Similar devices are devices that fall within the same general device category. The MDR defines this as a group of devices with the same or comparable intended uses or a shared technology that enables them to be categorized in a general way without reflecting characteristics.
What are the available guidelines and guidance documents for clinical evaluation?
The amended ‘Blue Guide’ on the application of the product rules 2022’ (“Blue Guide”) was released by the European Commission on June 29, 2022.
The Blue Guide allows a better understanding of EU product regulations and their uniform and coherent application across various sectors throughout the EU single market.
The Blue Guide has undergone significant changes, including the definition of new terms, the addition of information on which economic actors will be responsible for compliance in a complicated product supply chain, and the incorporation of Regulation (EU) 2019/1020 on market surveillance and product compliance.
Technical Documentation of The ‘Blue Guide’
The manufacturer must compile the technical documentation, including details proving the product complies with all relevant specifications.
If the law mandates a conformity assessment process based on a quality system, this paperwork may be a component of the quality system documentation.
Regardless of the product’s origin or location, technical documentation must be available when the product is put on the market.
The technical documentation must be preserved for ten years following the date the product was placed on the market. The manufacturer or the authorised representative based in the Union oversees this.
The documentation must include
Description of the product
Intended use of the product
Design and manufacture of the product
Operation of the product
The requirements in Annex II of Decision No. 768/2008/EC concern the technical documentation necessary to demonstrate the product’s compliance with the relevant harmonisation legislation.
If only part of the harmonised standard is applied or does not cover all relevant essential requirements, then the way applicable essential requirements not covered by it are dealt with should be documented in the technical documentation.
The technical documentation must reflect all versions of the product, including the changes made, information on how the various conformity assessments can be identified, and information on how the different versions of the product can be identified to avoid scenarios in which, during a product’s life, a market surveillance authority must deal with product versions for which the technical documentation given to it does not apply.
Even if it isn’t explicitly stated in the Union harmonisation legislation, the documentation must always be in a language the notified body can understand.
EU Declaration of Conformity
As part of the conformity assessment process outlined in the Union harmonisation legislation, the manufacturer or the authorised representative formed within the Union must prepare and sign an EU Declaration of Conformity.
This document is required to show the product’s compliance with the applicable legislation requirements.
Unless the legislation specifies otherwise, the manufacturer must maintain the EU Declaration of Conformity for 10 years after the product is placed on the market. The importer is accountable for the Declaration of Conformity for products they have brought in.
It is necessary to keep the EU Declaration of Conformity updated. Even if they are produced in series, each product has its own EU Declaration of Conformity.
The version of the EU declaration of conformity must be updated for products put on the market after any modifications have been made to any elements of the EU declaration of conformity.
Either the model declaration found in Annex III of Decision No. 768/2008/EC or a model declaration directly annexed to the in question sectoral Union harmonisation legislation have to be referred to understand the contents in the EU Declaration of Conformity.
The declaration must include enough details to allow the identification of all the products it covers, whether in the form of a document, label, or equivalent.
To ease the administrative burden on economic operators, where multiple pieces of Union harmonisation legislation apply to a product, the manufacturer or the authorised agent must produce a single declaration of conformity.
The surveillance authority must access the EU declaration of conformity upon request. The declaration must always be made in the language(s) that the Member state(s) where the product is marketed requires.
Marking requirements
Before many products may be marketed on the European market, a CE Mark must be affixed to them. The label identifies a product as:
Complies with the relevant standards of European product directives
Satisfies all requirements outlined in Europe’s applicable, recognised, and harmonised performance and safety standards.
Appropriate for its intended use and won’t threaten people or property
The CE Mark is mandated conformity marking used by the European Union (EU) to control the sale of goods inside the European Economic Area (EEA).
A manufacturer certifies that their products conform with the EU’s New Approach Directives by placing the CE mark on them. These directives include products made in or intended for sale in the EEA and those sold in the EU. As a result, the CE symbol is identifiable everywhere, even by those unfamiliar with the EEA.
The manufacturer is ultimately in charge of the product’s compliance with the provisions of the Union harmonisation legislation and the use of the CE marking, regardless of whether they are based inside or outside the Union.
The manufacturer has the right to direct an authorised agent to apply the CE marking on his behalf. By placing the CE marking on a product, a manufacturer certifies that it complies with all applicable regulatory requirements for CE marking, on his sole responsibility.
Suppose the importer or distributor or another operator places products on the market under his name or trademark or modifies them. In that case, he then takes over the manufacturer’s responsibilities, including the responsibility of affixing the CE marking.
The definition, the format, and the general guidelines governing the CE marking are outlined in Regulation (EC) No. 765/2008. Procedures for conducting conformity assessments that result in its affixing are outlined in Decision No. 768/2008/EC.
The Regulation (EC) No 765/2008 and Decision No 768/2008/EC’s guiding principles are primarily upheld by the sectoral Union harmonisation legislation requiring the application of the CE marking.
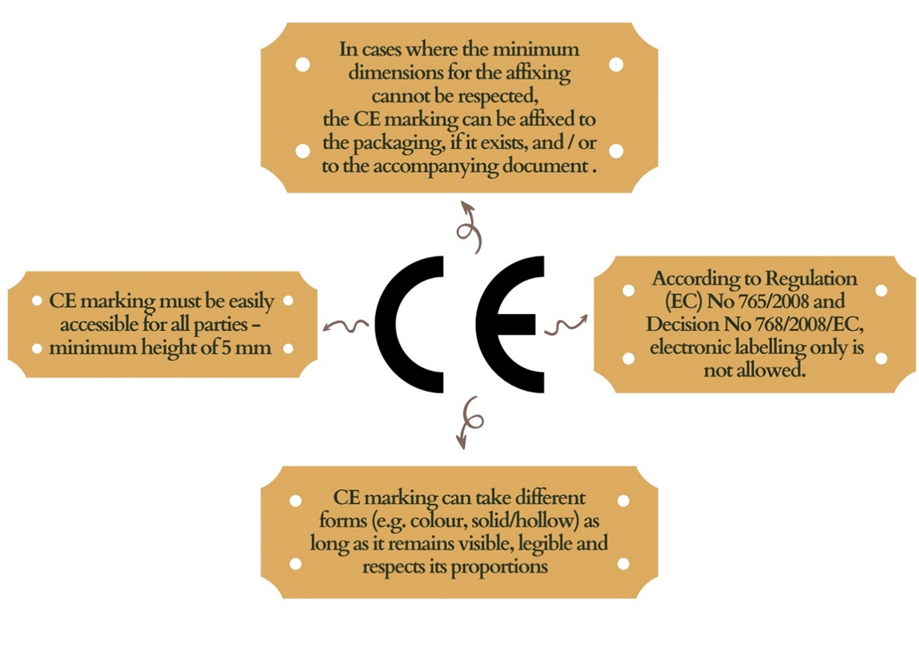
If a notified body participates in the production control phase following the appropriate Union harmonisation law, its identification number must come after the CE marking.
If the legislation so demands, the manufacturer or the authorised agent must attach the identifying number under the supervision of the notified authority.
A notified body may participate in the production stage depending on the conformity evaluation techniques. Only if it engages in manufacturing must the notified body’s identification number come after the CE marking.
CE marking appears on products without an identification number
CE marking appears on products with an identification number
Either no notified body intervened in the design or production phase (module A)
Either upon manufacturer’s choice, a notified body intervened in the production phase (modules A1, A2)
Upon manufacturer’s choice, the in-house accredited body intervened in the production phase (modules A1, A2)
A notified body intervened in the design phase (module B), and upon the manufacturer’s choice, a notified body (not necessarily the same one but the one whose identification number appears) intervened in the production phase (modules C1, C2 following module B)
A notified body intervened in the design phase (module B), but no notified body intervened in the production phase (module C following module B);
A notified body intervened in the design phase (module B), and a notified body (not necessarily the same one but the one whose identification number appears) intervened in the production phase (modules C1, C2, D, E, F following module B)
A notified body intervened in the design phase (module B), and upon the manufacturer’s choice, the in-house accredited body intervened in the production phase (modules C1, C2 following module B)
A notified body intervened in the design and production phase (modules D1, E1, F1, G1 H, H1)
Modules for Conformity Assessment
A conformity assessment is any procedure by the manufacturer to evaluate a product, system, service, or perhaps even a person’s compliance with the standards and specifications outlined in a standard or specification.
Testing or inspection is frequently used for verification. Conformance assessments are performed on products during the design and manufacturing phases.
A conformity assessment procedure’s primary goal is to show that products that have been put on the market adhere to the standards set out in the existing legislation.
Conformity assessment processes comprise one or two conformity assessment modules under Union harmonisation legislation. A conformity assessment encompasses both the design and production phases since products are subject to conformity evaluation during both phases.
In contrast, a module may cover just one of the two phases or both. A “horizontal menu” of conformity assessment modules and how processes are constructed from modules is outlined in Decision No. 768/2008/EC.
Union harmonisation legislation creates conformity assessment processes either by foreclosing on the manufacturer’s options or by defining a range of options from which the manufacturer must select.
The manufacturer is responsible for conformity evaluation. However, a third party must be included in the compliance evaluation process if required by the applicable legislation.
FAQs
What is the significance of CE marking?
By applying the CE marking to a product, the manufacturer declares solely on his responsibility that the product complies with the essential requirements of the applicable Union harmonisation legislation requiring its application and that the relevant conformity assessment procedures have been completed. Products bearing the CE mark are presumed to comply with the applicable Union harmonisation legislation and thus have free circulation in the European Union.
Can I, as a manufacturer, personally affix the CE marking to my products?
After the required conformity assessment procedure has been completed, the manufacturer or his authorised representative can apply the CE marking. This means that the product must go through the conformity assessment procedure outlined in one or more of the relevant Union harmonisation acts before being given the CE marking and put on the market. The latter determines whether the manufacturer himself may conduct the conformity assessment or whether the involvement of a third party (the notified body) is necessary. The published ‘Blue guide’ helps product manufacturers understand how to place their products in conformity with the applicable product regulation.
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.
All medical devices are associated with inherent risks of some level. It is imperative to understand the medical device’s specific risks to a patient. Under EU MDR 2017/745, risk management is a continuous and iterative process.
Manufacturers are expected to plan, document, and implement risk management strategies in this process. These strategies may either eliminate the risk or mitigate the overall severity of the risk.
Medical Device Risk- Definition
As per Article II of EU MDR 2017/745, medical device risk is defined as ‘the combination of the probability of occurrence of harm and the severity of that harm’.
According to the definition, the strategies help prevent particular harm or risk and prevent severe harm.
Risk Management under MDR
Annex I section 3 of EU MDR 2017/745 mentions its requirements specific to the European medical device regulations. Manufacturers, under MDR, must implement the following aspects of risk management to be fully compliant.
Establish and document its plan for each device
Identify the known and foreseeable hazards associated with the device
Estimate and evaluate the risks associated with, and occurring during, the intended use and during reasonably foreseeable misuse
Eliminate or control the risks
Evaluate the impact of information from the production phase to the post-market phase on hazards and the frequency of occurrence of associated risks, the overall risk, benefit-risk ratio, and risk acceptability
Amend risk control measures if necessary
While implementing risk control measures to design and manufacture devices, the following aspects must be considered. Manufacturers must:
Eliminate risks through safe design and manufacture of the device
Take adequate protection measures (such as including alarms) if the risks cannot be eliminated
Provide information for safety (warnings/precautions/contra-indications) and training to users.
Certain medical device risks may be due to device usage errors. In Annex I Chapter I, MDR clearly states that such risks can be prevented by:
Reducing risks related to the ergonomic features of the device and the environment in which it is intended for use
Consideration of technical knowledge, experience, education, training and use environment, and the medical and physical conditions of intended users
How are device risks managed?
Risk management can be considered a 5-step procedure.
Step 1: Risk management plan
All these activities must be planned. The plan lays forth a strategy for risk management activities to be carried out throughout the product lifecycle.
This plan is documented in a risk management file containing the risk management plan and a risk management report.
Step 2: Risk assessments
Risk assessments evaluate the risk identified in normal and abnormal medical device use. Normal use of a medical device is the intended application of the device following all instructions by the manufacturer.
In contrast, abnormal use is when the medical device was used, violating the device instructions.
Step 3: Risk Control
Risks are controlled by implementing its plan. The risk-control measures chosen must be executed, and their effectiveness must be validated. This is done for an effective quality management system.
Step 4: Evaluation of residual risks
Complete elimination of risk may not be possible all the time. Therefore, it is imperative to identify the residual risk so that small and expected rather than massive, unexpected risks.
Step 5: Risk management review
As risk management is an iterative process, reviewing the risk control measures adopted and their effectiveness is imperative. This is ensured by post-market surveillance systems, clinical evaluation, and vigilance systems.
Maintaining updated risk systems and documents constitutes an effective quality management system for any medical device.
FAQs
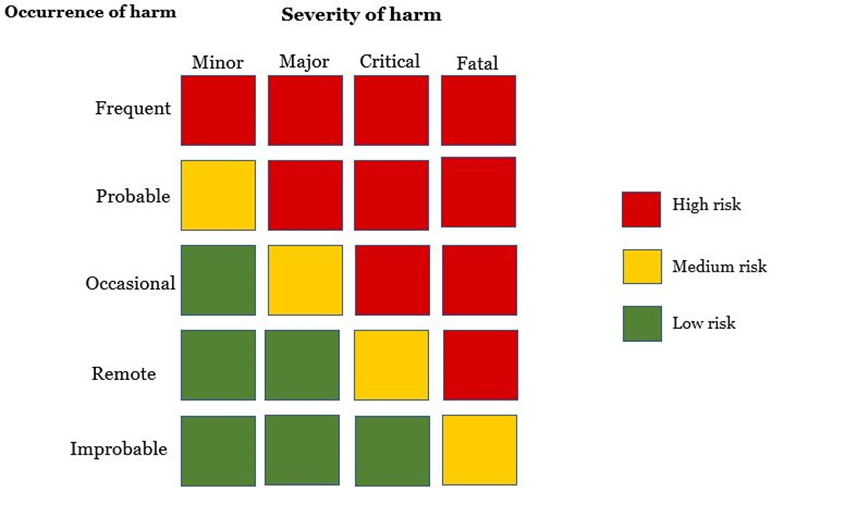
How are risks categorised?
Risks are classified based on the occurrence and severity of harm caused. The figure below is a risk matrix used to illustrate a matrix on all foreseeable risks. This is useful for evaluating residual risks posed by the medical device on the patient.
What is the EU MDR harmonized standard adopted for Risk Management?
EU MDR has adopted ISO 14971 for the Application of risk management to medical devices. This ISO standard allows manufacturers to identify hazards of a medical device and implement control measures for the same.
What is the role of Risk management in a clinical evaluation procedure?
Clinical evaluation is imperative to risk management as this allows the manufacturer to identify all possible risks associated with the device. This data can be used for the identification of safety concerns and appropriate risk management methods can be implanted. In other words, clinical evaluation is one of the inputs to risk management.
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.
The revolution in the digital sector has resulted in the Internet of Things (IoT), Software as a Medical Device (SaMD), Internet of Medical Things (IoMT) and other connected devices permeating the healthcare environment, both in hospital and home, has ended up with the possibility of cyberattacks and intrusions against the connected medical devices and the networks to which such a device is connected.
Most Medical devices are connected to the Internet, hospital networks, and other medical devices to provide health care and increase the ability of healthcare providers to treat patients.
These features also increase potential risks for Cybersecurity. Medical devices, like other computer systems, are vulnerable to security breaches, potentially impacting the safety and effectiveness of the device.
Since 2005, the FDA has tried to accomplish and enhance medical device cybersecurity, and the latest FDA effort is to create draft guidance that expects security throughout the total product life cycle (TPLC).
Another effort is the Protecting and Transforming Cyber Health Care Act of 2022 (PATCH Act of 2022),which, if passed, would revise the existing Federal Food, Drug, and Cosmetic Act.
The FDA guidance establishes six broad expectations on the Secure Product Development Framework (SPDF), which covers all aspects of a product’s life cycle, for the development, release, support, and decommission and satisfy Quality System Regulations (QSR) under 21 CFR Part 820:
Cybersecurity is a fundamental part of device safety and the QSR
Security by design
Transparency
Security risk management
Security architecture
Testing/objective evidence
The FDA draft guidance, under QSR, also declares that verification and validation activities by the medical device manufacturers shall include sufficient testing performed on the Cybersecurity of the system, which validates their inputs and outputs.
Further, the FDA summarizes that cybersecurity controls require testing beyond standard software verification and validation to demonstrate that the device has a good assurance of safety and effectiveness.
The following cybersecurity testing and corresponding objective evidence would be considered as the minimum support for a premarket submission:
Security requirements
Evidence of their boundary analysis creates a rationale for their boundary assumptions.
Threat mitigation
Evidence that all the design input security requirements were implemented successfully
Evidence of testing their threat models that demonstrates effective risk control measures provided in the system and use case
Evidence of the adequacy of risk control.
Vulnerability testing – Evidence on the testing of malformed
Abuse case and unexpected inputs
Vulnerability chaining
Closed box testing of known vulnerability scanning
Software composition analysis of binary executable files
Static and dynamic code analysis
Penetration testing– Identify and characterize security-related issues that discover security vulnerabilities in the product.
Regular interval cybersecurity testing – It is performed at regular intervals to identify the potential vulnerabilities before exploitation
This draft guidance replaces the 2018 draft version, which emphasizes the importance of understanding that all medical devices are designed securely, enabling new cybersecurity risks to be mitigated throughout the Total Product Life Cycle, and it elaborates the outline of the FDA’s recommendations more clearly for premarket submission to address cybersecurity concerns.
03/08/2022 Cybersecurity Alert: Vulnerabilities identified in medical device software components: PTC Axeda agent and Axeda Desktop Server
The PTC Axeda agent and Axeda Desktop Server are cloud-based technologies that allow people to securely view and operate the same desktop through the Internet. The Axeda agent and its desktop server are owned by the computer software company PTC.
The FDA alerts all medical device users and manufacturers about a cybersecurity vulnerability identified for the Axeda agent and Axeda Desktop Server.
The agent and desktop server of Axeda are used in many medical devices across several medical device manufacturers, and all the versions of the Axeda agent and Axeda Desktop Server are affected.
On the 8th of March, 2022, the Cybersecurity and Infrastructure Security Agency (CISA) published an advisory, ICSA-22-067-01, on these vulnerabilities.
Any successful exploitation of this vulnerability could allow an unauthorized attacker to take complete control of the host operating system, resulting in full system access, remote code execution, reading or changing the configuration, system file access, accessing log information, and other denial condition.
These vulnerabilities may result in changes to the functions of the medical device and impact the availability of the remote support functionality.
As a result, PTC recommends that affected manufacturers:
To upgrade Axeda agent Version 6.9.2 build 1049 or 6.9.3 build 1051 while running older versions of the Axeda agent.
Also, to configure the Axeda agent and Axeda Desktop Server to listen only on the local host interface 127.0.0.1.
Then, Provide a unique password in the AxedaDesktop.ini file for each and every unit.
Remove the installation file.
Make sure to delete the ERemoteServer file from the host device.
Never use ERemoteServer in production.
When running the Windows operating system, first configure Localhost communications (127.0.0.1) between ERemoteServer and Axeda Builder.
When running in Windows or Linux, only allow connections to ERemoteServer from trusted hosts and block all others.
Configure the Axeda agent for the authentication information required to log in to the Axeda Deployment Utility.
So, Cybersecurity is one of the crucial aspects of today’s fast pacing digital world. The threats caused by Cybersecurity, especially on medical devices, are hard to deny. It is important to learn how to defend themselves from them and create a safe environment for the usage of medical devices.
EU MDR and IVDR
In the EU, both the MDR and IVDR Annex I create requirements for mandate consideration of medical device cybersecurity, and the Medical Device Coordination Group (MDCG), in its guidance, explains to the manufacturers of medical devices how to fulfil all the relevant essential requirements regarding Cybersecurity.
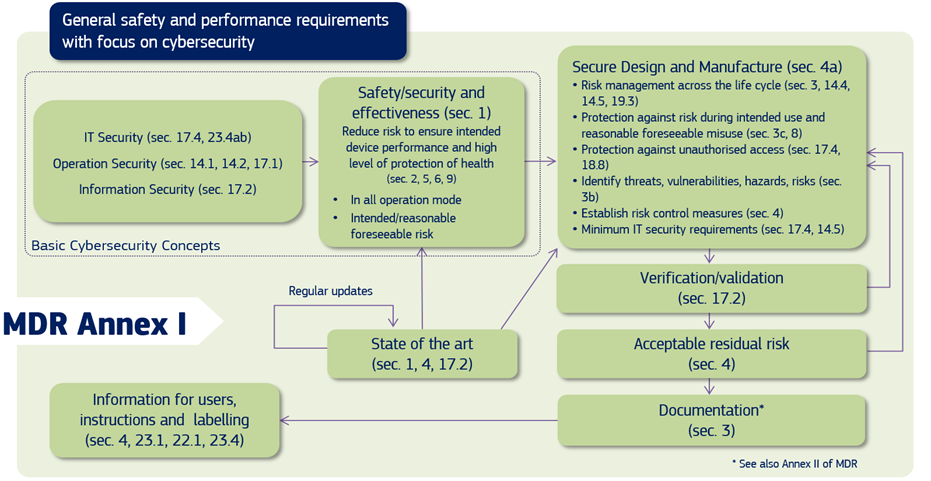
Source: MDCG 2019-16 Guidance on Cybersecurity of medical devices Figure 1: Cybersecurity requirements contained in MDR Annex I
The NIS Directive also provides for legal measures to increase the overall level of Cybersecurity in the EU.
GDPR (General Data Protection Regulation) helps the manufacturers of medical devices in regulating, protecting and processing personal data by the individual, company or organization that relates to the EU.
The EU Cybersecurity Act certifies Cybersecurity for ICT products, services, and processes.
According to the Cybersecurity Act, manufacturers are required to demonstrate state of art in the design, development, and improvement of their medical devices throughout their life cycle.
During that period, the manufacturers must consider the safety, security, and efficacy of the medical devices, and in vitro diagnostic safety mechanism design must be considered early during the manufacturing process.
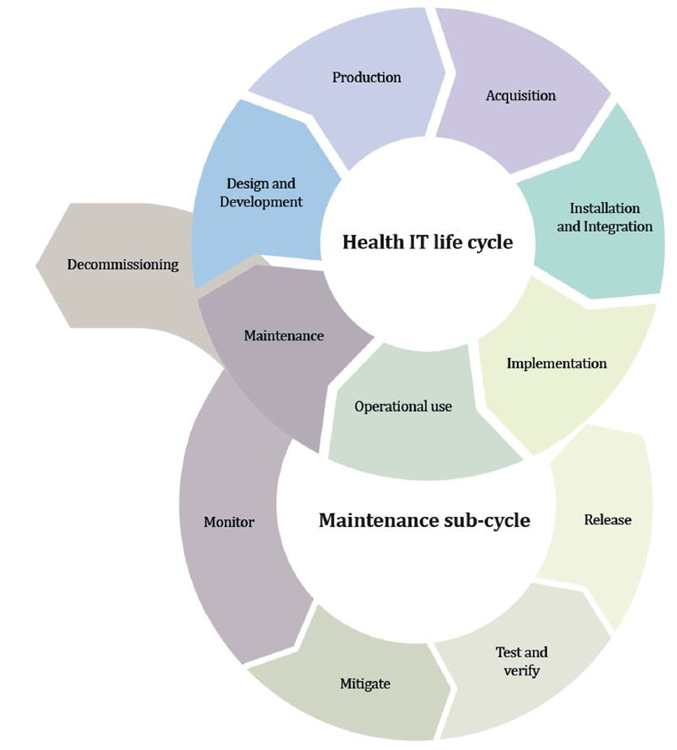
Source: MDCG 2019-16 Guidance on Cybersecurity of medical devices Figure 4: Lifecycle stages
The MDCG has proposed some key philosophies of the staged security concept strategy (“defense in depth strategy”) as follows:
Security management
Specification of security requirements
Security by design
Secure implementation
Management of security-related issues
Security update management
Security risk management
The list of possible IT security requirements for the operating environment according to MDCG:
Compliance with national and EU regulations (e.g., GDPR).
Ensuring appropriate security controls are in place
Ensuring the physical security of the medical device through security measures
Ensure control and security of network traffic through proper measures
Life Cycle Aspects
Security measures specific to their workstations connected to the medical device.
Security vulnerabilities related to the device hardware/software and third-party hardware/software used with the medical device.
During the life of the devices, the manufacturer should implement the process to collect post-market information about the security of the device.
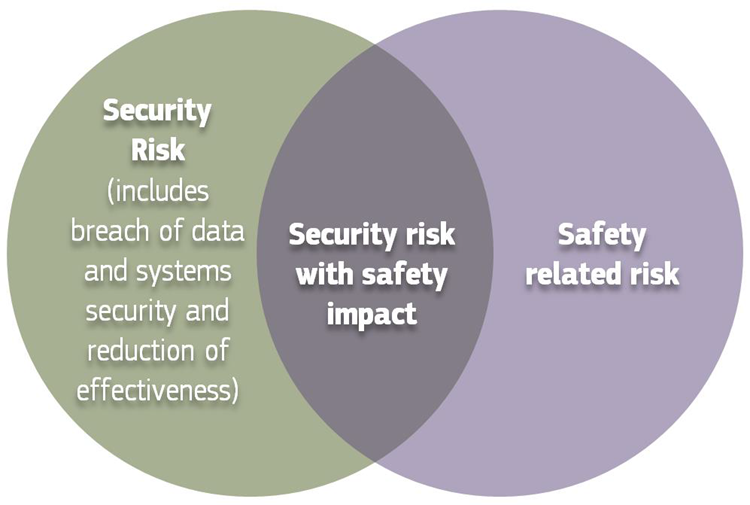
Source: MDCG 2019-16 Guidance on Cybersecurity of medical devices Figure 3: Cybersecurity measures may cause safety impacts
Based on the EU Cybersecurity Act, the manufacturer must provide the following information to the user of the medical device:
Specifications of the operating system
IT security risk assessment information.
Provisions for ensuring the integrity of software updates and security patches
Product installation
Security configuration options
Initial configuration guidelines
Step-by-step instructions for deploying security updates
Description of the backup and restore functions for data and configuration settings
Procedures for using all the medical devices in failsafe mode
The manufacturers are required to establish a post-market surveillance (PMS) system and actively keep these PMSs (Post Market Surveillance) up to date. Medical device cybersecurity requirements should be part of this PMS system.
Depending on the class of medical device, a PMS report or PSUR report will be generated, which concludes the analysis of all data from the market.
FAQs
How can we protect heath care from cyber-attacks?
· Vulnerability assessment and required testing · Training health care providers to protect from any breaches · Follow the standards of the regulations
Where is Cybersecurity used?
Cybersecurity helps in protecting the Datas, software or hardware connected with the system. This reduces unauthorized access to the data or the system.
What is the PATCH act?
PATCH act helps to meet all the Cybersecurity requirements for the manufacturer to complete FDA regulation standard.
What medical devices can be hacked?
MRI, Pacemakers, Implants, Heart rate monitors, Drug infusion pumps, medical records and other devices connected to the hospital network.
What are the new cybersecurity requirements according to EU MDR?
MDR Annex I explain the risks associated with the interaction between software and medical devices. Manufacturers should follow standard during life cycle, risk management, verification, and validation of the devices.
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.