A short article based on MDCG 2021-22 guidelines on “first certification for the type of devices and the corresponding procedures to be followed by the Notified Bodies, ” established based on Article 106 of Regulation 2017/745 for Class D IVDR devices.
Additionally, this guide talks about the conditions to be followed by the Notified bodies, whether to consult the expert panel for the performance evaluation report of the IVDR devices.
If the type of the device comes under the first certification in the market (AND)
There is no CS for the class D devices.
According to Article 48(6) of IVDR 2017/746, the first certification for the type of device means that the product has no similar product with the same intended use or technology around the European market.
Also, the notified bodies should submit the performance evaluation report to the expert panel within five days of receiving it from the manufacturer. The same intended purposes include
Device functions like monitoring, diagnosing, screening etc.
If the device is quantitative, semi-quantitative or qualitative
It is automated or semi-automated, or not
If it is used for any measurement
If the device is used for any specific disorder
What is the specimen type, or if it is applicable for some testing purpose
What all the intended users
Or if the device has the same principle of operation
Suppose the IVDR devices has met the above-mentioned purposes under Directive 98/79/EC or has an ongoing consultation with the expert panel under EU 2017/746. In such case, the notified bodies need not consult the expert panel for the certification.
However, if the class D device is with similar intended purpose but one of the IVDR devices has some additional intended purpose along with the common intended purpose, which falls outside Rule 1 or 2 of IVDR Annex VIII then, this type of device should be considered under the “type of device” while consulting the expert panel.
Other examples for the same type of purposes include
Automated and semi-automated devices
Technologies
Multiplex and single analyte devices
Kits and class D components of those kits
Control materials
Hence, the notified body issuing the certificate should check if the product meets the criteria stated above to classify it as the “type of device” for the first time.
For the final decision, they can use their knowledge and expertise, consider the information provided by the manufacturer, or if the device has already undergone an expert panel consultation.
Post assessments, if the notified bodies decide that the product is given the first certification, they need to consult the expert panel and document the certification with the conclusion.
Fig 1: Notified body template for the expert consultation
If the expert panel has been kept in consultation for the first certification of the IVDR devices, the notified body cannot issue the second or third certificate without considering the views of the expert panel on the type of device.
What are class D IVDR devices?
Class D devices are high-risk devices, including devices used for blood screening for grouping, to screen cells & organs to manage infectious outbreaks or for transplantation.
What is an expert panel?
Expert panel advisors are appointed by the EU commission who can provide scientific, technical, and clinical evaluation guidance for high-risk devices and their performances.
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.
Market surveillance is the activity carried out by the competent authorities according to Article 93 of MDR 2017/745 to ensure that products on the market comply with the relevant laws and regulations and with the current EU health and safety requirements. Maintaining market security in Europe and promoting consumer safety and business trust is vital.
Suppose the medical device poses an unacceptable risk to the health or safety of the patients, users, or other people or any other aspects of the protection of public health.
In that case, the competent authorities shall immediately require the manufacturer of the medical devices concerned, its authorized representative (AR), and all other relevant economic operators to take appropriate and duly justified corrective actions to bring the device into compliance with the MDR Regulation or withdraw or recall the medical device from the market.
Depending on the severity of the incident, the regulatory authorities require reports of serious adverse events or accidents to be submitted within 48 hours to 15 days.
The most crucial aspect of market surveillance efforts is the evaluation and examination of the adverse event or incidence investigation reports, risk management reports, and vigilance data from medical device manufacturers and consumers.
If they disagree with the national legislation notified, they must immediately communicate their complaints to the European Commission and the other Member States via the electronic system referred to in Article 100 of MDR.
Any measures taken by a Member State that has not been objected to by either a Member State or the Commission within two months of receiving the notification are justified.
If necessary, the authority may start a public field safety corrective action (FSCA). For significant product non-conformities, the responsible authority must evaluate if further regulatory action is necessary, which can be taken by destroying hazardous or non-compliant products, confiscating medical equipment, or assisting the manufacturer in bringing the product back into compliance with the law.
Obligations of the competent authorities are
Monitoring devices and incidents that occur while using them
Analyzing market data and identifying the possible potential issues
Reviewing technical documentation
Identifying unsafe or non-compliant medical devices
Reactively or proactively inspecting economic operator facilities
Removing unsafe devices from the market
The competent authorities shall develop annual surveillance activity plans and allocate adequate materials and qualified human resources to carry out those activities above.
Also, they shall prepare a summary of the surveillance activities performed with the required corrective action to be set out by the economic operators and make them available on the electronic system.
The Member States will review and evaluate the effectiveness of their market surveillance efforts, which are carried out at least every four years, and be informed to the Commission and other Member States of their outcomes.
Each Member State must summarise the results available to the public via the electronic system, as mentioned in Article 100 of MDR 2017/745.
The electronic system EUDAMED shall collate and process the following information on market surveillance.
Surveillance activity results
Final inspection report
Unacceptable risks associated with the medical devices
Non- compliance product information
Results of reviews and assessments carried out by the Member states on market surveillance.
To ensure a high-quality and uniform level of market surveillance across all Member States, the competent authorities must coordinate their market surveillance efforts through joint market surveillance activities, work together, and share their results with one another and the Commission.
What is the difference between Post-market Surveillance (PMS) and Market Surveillance?
Market surveillance is the same as post-market monitoring, where they make sure that medical devices sold on the EU market are safe, effective, and compliant. Market monitoring is conducted by the appropriate competent authorities, whereas the manufacturer of the medical devices carries out PMS.
What is the role of vigilance in market surveillance activities?
Vigilance focuses on reporting serious adverse events or accidents and field safety corrective actions (FSCA). In contrast, market surveillance is entirely concerned with developing procedures that can ensure the compliance of medical devices.
Any serious incident involving a device marketed and sold in the European Union and any field safety remedial action carried out in the EU or other markets where the device is sold should be made available in EUDAMED.
What is a joint action of market surveillance?
The Joint Action on Market Surveillance of Medical Devices (JAMS) is created to strengthen market surveillance across European member states. The medical device industry carries out best practices, education, information, and resources to improve public health protection. Critical goals include enhancing coordination and assisting member states with limited resources in building their capacities and skills in the market surveillance network. It will facilitate the creation of clinical processes, equivalent resources, and a consistent and balanced approach to manufacturer inspections.
Article 11 of the Regulation (EU) 2017/745 on medical devices (MDR) and EU Regulation (EU) 2017/746 on in vitro diagnostic medical devices (IVDR) outlines the obligations and responsibilities for the Authorised Representatives (AR).
Key points are taken for Authorised Representatives in accordance with the guidance:
A ‘sole’ authorised representative means a manufacturer can have more than one Authorised Representatives (AR) for different generic device groups. However, for one specific generic device group, the manufacturer should designate only one Authorised Representatives (AR).
On behalf of the manufacturer, Authorised Representatives (AR) can lodge an application for conformity assessment according to Annex IX, X, XI MDR/IVDR with a notified body at the pre-market stage.
AR who terminates the mandate may still cooperate with the competent authority for devices which was designated by them, particularly when no new AR has yet been designated for those devices.
AR must be registered under EUDAMED as per Section 1 of Part A of Annex VI of the Regulations, which needs to be updated within one week of any change occurring and the accuracy of the data at intervals no later than one year after the initial submission of the information, and every second year thereafter.
AR name shall appear on relevant documentation such as the EU DoC (Declaration of Conformity) and device labelling by the manufacturer.
The legal representative established by the sponsor of the clinical investigation or performance study is not defined under the AR requirements.
AR shall keep a copy of the technical documentation and the EU declaration of conformity for at least ten years after the last device covered by the EU DoC and 15 years for implantable devices.
‘Permanently available’ means it is mandatory for the manufacturer to provide the requisite documentation in their most recent versions and certificates. This includes amendments or supplements, either in hard or electronic copy to the AR. In practical terms having permanent access to such documents.
When the liability of the AR is alleged within the framework of a specific legal regime for defective products, the AR is afforded the same rights to defend itself as the manufacturer under that regime.
In the event of a problematic termination like non-compliance or non-traceability with the manufacturer, the out-going AR is also advised to inform the competent authorities and, where applicable, the notified body.
In general, a tripartite agreement should exist, except in cases where this is ‘not practicable’ to involve the outgoing AR, the obligation under Article 12(d) to forward complaints to the manufacturer or incoming AR should apply.
AR may be consulted for market surveillance measures taken by competent authorities to make documentation and information available to them. Also, AR may be subject to announced and unannounced inspections by the competent authorities as part of their market surveillance activities which is also covered for the legacy devices.
New guidelines on how to distinguish between medical devices and medical products under the Medical Devices Regulation have been published by a working group of the European Commission (MDR).
The new recommendations from the European Medical Device Coordination Group (MDCG) cover “borderline products” that are difficult to classify as either medical devices subject to the MDR requirements or medical products for human use subject to the requirements of Directive 2001/83/EC (MPD) for CE Marking.
The agreements reached by the Member State members of the Borderline and Classification Working Group (BCWG) following the exchanges under the Helsinki Procedure in accordance with Regulation (EU) 2017/745 on medical devices (the MDR) and Regulation (EU) 2017/746 on in vitro diagnostic medical devices are explained in the document, which will be referred to as the manual (the IVDR).
The specific document here explains the goals and workings of the Helsinki procedure. The BCWG is presided over by the European Commission and comprises observers from a number of stakeholder associations and representatives of all Member States’ relevant agencies.
The document outlines the methodology for classifying medical devices per the new regulatory framework. At the same time, the document itself is not legally binding and is not meant to establish new regulations or responsibilities. Instead, it further explains the relevant regulatory requirements and suggests considerations for ensuring compliance.
It is clearly emphasised that the current document is not intended to represent the EC’s official viewpoint. Additionally, should it be deemed reasonable to do so to reflect comparable adjustments to the underlying rules, the MDCG maintains the right to make changes to the document and suggestions included within.
Regulatory Background
As a general rule, it is the responsibility of the authorities of the Member States where a product is on the market to determine whether it qualifies as a medical device and to apply the classification requirements.
The MDCG also recognises that there may be variations in the methods used depending on how the applicable classification rules and regulations are interpreted. The MDCG believes it is crucial to create a widely followed protocol that will be adhered to by all parties engaged in activities involving medical devices in the EU.
The agreement that the Member States reached following their interactions under the MDR and IVDR of the Helsinki Procedure is described in the manual.
Scope
The manual defines borderline cases as situations where it is not immediately evident whether a particular product is a medical device, an in vitro diagnostic medical device (IVD), or neither.
Additionally, it is indicated that some of the requirements found in other articles of the Regulations, including those that list the products exempt from the Regulations’ purview, may also be applied.
However, in some circumstances, even if the product in question does not fit the definition of a medical device or is expressly exempt from regulation under the regulation, other EU-wide rules or national regulations may still apply.
These situations are not covered by the present manual, which only covers the aspects of using the qualification rules outlined in the Regulations.
The document also refers to the MDCG guidance on the distinction between medical devices and pharmaceuticals under the Medical Devices Regulation (EU) 2017/745, as well as the relevant guidance on the qualification and classification of software under Regulations (EU) 2017/745 and 2017/746.
The relevant regulations further state that after it is established that the product in question is a medical device, a risk-based classification should be carried out, resulting in the device being given the proper risk class (from I to III).
The risk classes to be used are A, B, C, and D if the product under consideration is a medical device for in vitro diagnosis. As was previously indicated, under some circumstances, it becomes challenging for the competent authorities of the Member State to apply the requirements for medical device classification uniformly.
The competent authority should make its choice on a case-by-case basis, considering the details of the product in issue, as the suggestions offered in the current handbook are not all-inclusive.
Key Principles
Borderline Between Medical Devices and Medicinal Products
The following are only a couple of the instances given by the MDCG:
A nasal spray containing COVID-19 antibody is intended to render the virus inactive. Before anything else, it’s critical to identify the primary mode of action or how the product fulfils its intended function.
In this scenario, the main goal of the spray is accomplished by antibodies attaching to the virus; as a result, the virus cannot replicate and infiltrate mucosal cells.
Furthermore, it is said that a product that meets the general definition of a medical device is one whose intended action cannot be accomplished through pharmacological, immunological, or metabolic methods.
Accordingly, based on this criterion, the product in question should be controlled as a medicinal product rather than a medical device.
Graphite crucible used to create pictures of the patient’s airways using Technetium-99m radionuclide (to prepare an aerosol). While a gamma camera is used to capture an image, such an aerosol should be inhaled.
Any medicinal preparation that, when ready for use, has one or more radionuclides (radioactive isotopes) incorporated for medicinal purposes are radiopharmaceutical, according to the appropriate regulations. A kit is any preparation that must be reconstituted or mixed with radionuclides in the finished radiopharmaceutical, often before administration.
Due to the product’s intended use and planned mode of operation, it does not fulfil the definition of a medical device in this specific instance and is, therefore not subject to regulation under the medical device framework.
Borderline Between Medical Devices and Biocides
The guideline also explains the crucial distinctions between medical devices and biocides, governed by Regulation (EU) 528/2012, which deals with issues pertaining to biocidal goods intended for marketing and use in the EU.
The authority also offers many instances to illustrate the methodology used when making such a conclusion. For instance, an antibacterial and antiviral concentration is described in the first example as a water-based treatment for textile materials.
In this situation, the treatment in question does not fulfil the definition of a medical device. It should not be regulated as one because it is designed to affect the features of other items rather than apply to specific patients.
Other Types of Products
The guidelines scope also includes topics pertaining to substances with human origin, cosmetics, food, and personal protective equipment. A rescue bag is the first product that is described. It transfers patients during rescue operations to ensure their care and safety.
Such a bag, in particular, comprises components meant to guarantee the patient’s mechanical protection while transported, including various transport mechanisms. The main goal of the rescue bag is to prevent the patient’s condition from worsening due to any environmental influences during the rescue.
According to the authority, the product should be classified as a medical device because its intended use conforms to the medical purpose of easing or compensating for an injury or handicap, as stated in Art. 2(1) of the MDR; nonetheless, according to Rule 1, the risk class should be MDR class I.
Another illustration in the manual explains the Plexiglas box for caregiver protection, which is meant to be utilised to lower the dangers associated with the infection that could be spread during procedures.
In this regard, the authority explains that a product should not be classified as a medical device if its only purpose is to protect a caregiver or healthcare professional by preventing exposure during a medical or surgical operation.
Further, since a medical gadget is supposed to be used to safeguard the patient, such a motive could not possibly be its intended use.
FAQs
What does the Helsinki Procedure mean?
The Helsinki Procedure is a procedure that “allows consultation among competent authorities (CAs) on borderline and classification issues concerning medical devices and to guarantee that relevant guidance is provided in the Manual on Borderline & Classification for Medical Devices.
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.
This article focuses on the distinctions between the MDD and MDR, considering the terminologies and real-world examples of substance-based devices and combinations of medical and medicinal products.
The guidance distinguished the following three concepts in MDR.
1. Article 2(1) MDR, first paragraph refers to the ‘Specific medical purpose’ specified by the manufacturer as
diagnosis, prevention, monitoring, prediction, prognosis, treatment of disease
diagnosis, treatment, monitoring, alleviation of, or compensation for, disability or injury
the investigation, modification, or replacement of the anatomy or a physiological or pathological process or state
2. Article 2(1) MDR, second paragraph refers to ‘Principal intended action’, which describes the manufacturer’s labelling and claims based on state-of-the-art scientific data regarding the principal mode of action, on a case-by-case basis like
the medical devices for the control or support of conception
products specially intended for the cleaning, disinfection, or sterilization of devices
3. Article 1(6)(b) MDR refers to the ‘Principal mode of action’ by which the product meets its principal intended action, i.e., pharmacological, immunological, metabolic, physical, or other factors.
Substances-based medical devices
A medical device that uses substances as its base contains ingredients approved for use in medical devices and does not carry out its primary intended activity by pharmacological, metabolic, or immunological mechanisms.
Those devices are used equivalently as a medicinal product, for example, ingested or applied to the skin.
In addition to outlining broad guidelines for substance-based medical devices, the guidance offers an explanation and examples for the substances-based devices that fall under two rules according to MDR Annex VIII.
Rule 3 talks about the medical devices containing a chemical or combination of substances utilized in vitro in direct touch with human cells, tissues, organs, or human embryos before implantation or administration
For example
IVF or ART (Assisted Reproductive Technology) products without principal pharmacological/metabolic action (substances or mixture of substances)
IVF cell media without human albumin
Solutions for the transport of organs for transplantation (that do not achieve their principal intended action via pharmacological, immunological, or metabolic means),
Rule 21 talks about the medical devices made of substances or mixtures of substances that are meant to be injected into or applied to the skin
For example
Vaginal lubricants/moisturizing gel
Salt water used for nose or throat sprays
Skin treatment formulations
Eye drops for lubrication
Ear drop
Oral administration of medical devices to treat obesity
Medical Device and Medicinal Products combination
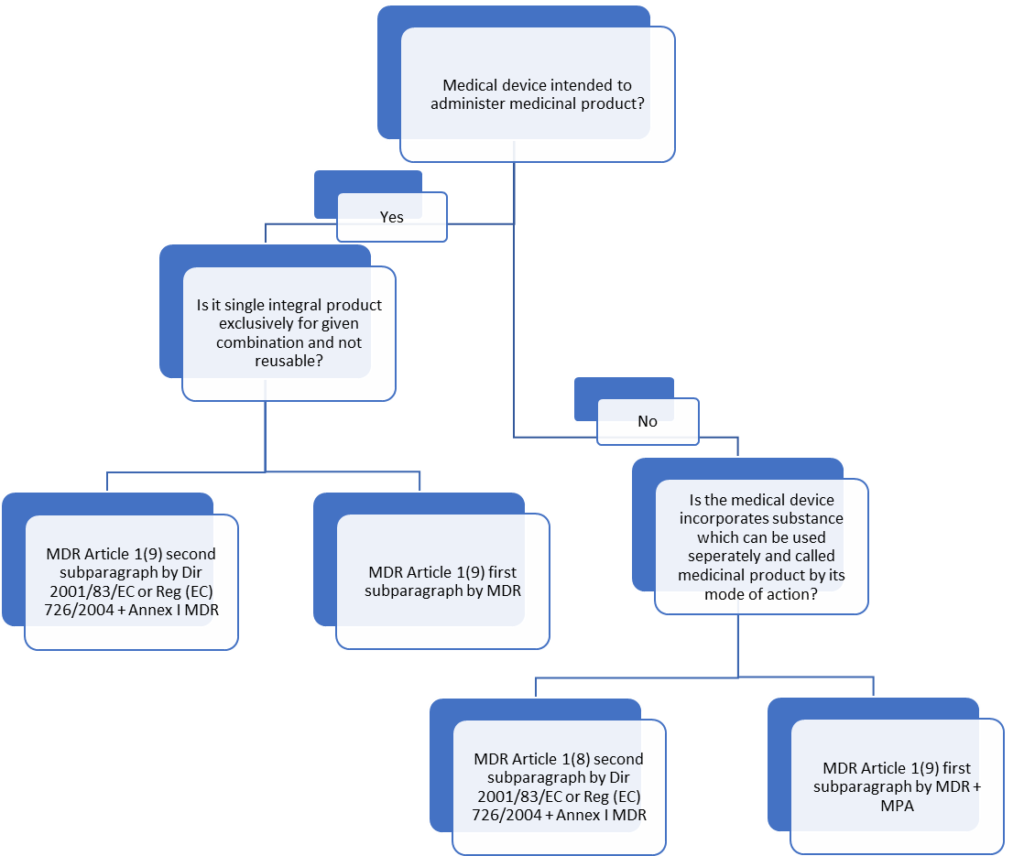
The MDR Article 1(8) and Article 1(9) give a helpful flow chart (Fig 1) for medical devices that are intended for use with a medicinal product in a combination product.
Flowchart to find the regulatory requirement of the combination product
What is meant by “integral” is the key concept in MDCG 2022-5 SECTION 4:
Article 1(8), First Paragraph of MDR, uses the phrase “integral part” to describe components of devices that would be classified as therapeutic items if used independently.
In the second paragraph of Article 1(8) MDR, the term “integral products” refers to devices that incorporate a material that, if taken separately, would be considered a medicinal product.
When a product is on the market, it must have at least two separate components, one of which is a device that, when connected (for example, physically or chemically), functions as a single unit and comes under integral product.
As an exceptional case, medical devices co-packed with a medicinal product or devices included in the information provided with the medicinal product are not considered integral products if the pertinent combination occurs at the time of administration.
Devices used to deliver a medicinal product, and the respective medicinal product forms a “single integral product” are mentioned in Article 1(9), second paragraph MDR.
A single integral product comprises at least two constituent elements, one of which is a device and the other is a pharmaceutical, mixed in a way that prevents their intended separation before use.
If the substance accomplishes the primary intended action of the integral product, the entire product is governed in accordance with Directive 2001/83/EC or Regulation (EC) No 726/2004 as a medicinal product.
If the medical device accomplishes the primary intended function, the complete product is governed by the MDR as a medical device containing a medicinal substance with an additional function.
Devices for administering medications, where the medication is provided separately, are not integral products.
MDCG 2022-5 provides clear definitions with examples for the following:
Combinations of medical devices and medicines are regulated as medicinal products.
The MDR states that Directive 2001/83/EC or Regulation (EC) No. 726/2004 governs the entire product but that the relevant general safety and performance standards of Annex I MDR shall apply as far as the safety- and performance-related device characteristics are concerned.
If the device has a CE mark, the conformity assessment results must be included in the marketing authorization dossier, including the variation dossier.
If the dossier does not contain this information, however, and if a notified body would be required to conduct a conformity assessment of the device if used separately, an opinion on the conformity of the device part with the general safety and performance requirements listed in Annex I MDR must be provided. For further guidance, refer to EMA (European Medicines Agency).
For example
Pre-charged nebulizers with the specific medicinal products
Pre-filled syringes with the specific medicinal product
Devices designed to administer a medicinal product within the meaning of the MPD fall under this category, even when the device and the medicinal substance are not integrated.
For example
Jet injectors
Port systems
Implantable infusion pumps
Drug delivery pumps
Spacers intended for meters dose inhalers
Medical devices with an ancillary medicinal product built in as a component
The MDR outlines the situation in which medical devices contain a substance that, if used alone, would be considered a medicinal product under Article 1 of the MPD, including a medicinal product made from human blood or human plasma, with an action unrelated to the device’s intended purpose which includes herbal medicinal products too.
For example
Bone void fillers containing growth factors
Condoms coated with spermicides
Liquid wound dressing containing the anti-microbial agent
Catheters coated with heparin or an anti-microbial agent
Medical devices that include herbal products
Clove Oil (Caryophylli aetheroleum) – has antiseptic, analgesic, and sedative properties.
Thymus vulgaris (Thyme) – disinfectant, antiseptic and expectorant properties
Medical devices that include human blood or its derivative
Culture media used in IVF containing human albumin solution
Haemostatic agent/matrix containing human thrombin
Annex VIII, Rule 14 states that ‘All devices incorporating, as an integral part of a substance which, if used separately, can be considered to be a medicinal product, as it is defined in point 2 of Article 1 of Directive 2001/83/EC, including medicinal product which derived from human blood or human plasma, as defined in point 10 of Article 1 of that Directive 2001/83/EC, and that has an action ancillary to that of the devices, are classified as class III’.
Furthermore, Annex IX section 5.2(a) requires that ‘Where a device incorporates, as an integral part of a substance which, if used separately, may be considered to be a medicinal product within the meaning of the point 2 of Article 1 of Directive 2001/83/EC, including a medicinal product that derived from human blood or human plasma and that has an action ancillary to that device, the quality, safety and usefulness of substance shall be verified by analogy with the specific methods in Annex I to Directive 2001/83/EC’.
FAQs
What is EMA?
The European Medicines Agency (EMA) is an agency of the European Union which regulates the evaluation and supervision of medicinal products. Its main responsibility is to protect and promote public and animal health.
What is a combination product?
Combination products are two or more components, such as a drug and a device, a drug and a biological, a biologic and a device, or a drug and a device and a biological, must be integrated, mixed, or combined in some other way to be considered a single product.
Is UDI (Unique Device Identification) applicable for medicinal products incorporating medical devices?
UDI-related MDR duties are not necessary for a DDC (Drug Device Combination) covered by the regulation of the medicinal products. Therefore, the package of such a DDC should not be subjected to a device part-related UDI. Additional information is provided in the MDCG 2019-2 guidance on the application of UDI rules to device-part of products referred to in Articles 1(8), 1(9) and 1(10) of Regulation 2017/745. When a DDC’s device component is CE-marked, the integral DDC’s product labelling should adhere to the labelling specifications for medicinal products as specified in the QRD (working group on Quality Review of Documents) templates. There is no need to remove a UDI that has already been physically marked on a device part. The labelling or outside packaging of the pharmaceutical product should not include the UDI.
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.
A notified body is an independent organisation designated by an EU country to assess the conformity of products before being placed on the market. Under EU MDR 2017/745, the notified body is also called a conformity assessment body.
A conformity assessment body (CAB) or a notified body is responsible for carrying out the conformity assessment procedure mentioned in the applicable Regulation.
What does a notified body do?
A notified body assesses whether the product conforms to the requirements set out in the legislation using a conformity assessment procedure. Some essential activities under the procedure include testing, certification, and inspection.
A notified body is a third-party organisation that works for the benefit of manufacturers. The list of Notified bodies is updated on the NANDO website (New Approach Notified and Designated Organisations).
A notified body also has a list of requirements that need to be fulfilled to carry out conformity procedures.
What is a conformity assessment?
Any legal manufacturer of a product can place their products if the product meets all the requirements. Before a product may be sold, it must undergo a conformity evaluation.
The primary goal of the European Commission is to help ensure that dangerous or otherwise non-compliant items do not enter the EU market.
A conformity assessment is done
Before a product is released on the market, its conformance is evaluated.
To demonstrate compliance with all regulatory standards, which include testing, inspection, and certification.
For each product, the applicable product regulation specifies the method of conformance.
In turn, a conformity assessment guarantees the following:
The product about to be placed on the market meets all legal standards.
The procedure ensures the confidence of consumers, public authorities, and manufacturers regarding the conformity of products.
How is the conformity procedure done?
Product legislation describes the conformity assessment method for each individual product. In the case of medical devices, EU MDR 2017/745 is the latest applicable legislation.
Manufacturers may choose between different conformity assessment procedures, if applicable. The manufacturer may carry out the assessment. If the appropriate legislation requires it, a conformity assessment body is involved in the conformity assessment process.
At the end of the conformity assessment, a CE certificate is issued, which implies that the medical devices conform to the requirements set out in the applicable Regulation.
This ensures that the manufacturer can freely market the device in the EU. The CE certificate and the device bear the CE marking adjacent to which the number of the notified body that issued the same is present.
CE marking with the number of the Notified body
Requirements to be met by Notified Bodies
Annex VII of EU MDR 2017/745 mentions the requirements for Notified bodies. Some general requirements like organisational structure, impartiality and confidentiality are mentioned. More specific requirements include the following:
Maintenance of an appropriate QMS
The Quality Management System must be established such that management structure, documentation, policies and objectives are clear. The QMS must ensure that good documentation practices are followed. This includes control of documents and records, continuous reviews and frequent audits.
Resource requirements
Notified bodies must be capable of carrying out all tasks assigned to them under the Regulation with the utmost professional integrity and competence in the specific field, whether done by notified bodies themselves or on their behalf and under their supervision. The personnel responsible for the tasks carried out by notified body must have suitable qualifications and adequate expertise.
Process requirements
The notified body must have documented processes and sufficiently detailed procedures in place for the conduct of each conformity assessment procedure for which it is designated, including the individual steps from pre-application activities to decision-making and surveillance and considering, as necessary, the respective device specification.
FAQs
Do all EU countries have a notified body?
No. Some countries do not have a notified body. The complete list of notified bodies under EU MDR is provided here. Some examples of notified bodies include TUV, SGS and BSI. These notified bodies do not have a presence in each European country, but the certification they provide is accepted elsewhere.
How do I choose the best-notified body for my devices?
While there is no one best-notified body, a couple of things should be considered when choosing one. Cost of the conformity assessment procedure, response, and geographic location of the notified body. Furthermore, choosing a notified body that also allows the MDSAP certification is considerable if the device is marketed to countries like the US, Canada, and so on.