


FDA –Food and Drug Administration
The revolution in the digital sector has resulted in the Internet of Things (IoT), Software as a Medical Device (SaMD), Internet of Medical Things (IoMT) and other connected devices permeating the healthcare environment, both in hospital and home, has ended up with the possibility of cyberattacks and intrusions against the connected medical devices and the networks to which such a device is connected.
Most Medical devices are connected to the Internet, hospital networks, and other medical devices to provide health care and increase the ability of healthcare providers to treat patients.
These features also increase potential risks for Cybersecurity. Medical devices, like other computer systems, are vulnerable to security breaches, potentially impacting the safety and effectiveness of the device.
Since 2005, the FDA has tried to accomplish and enhance medical device cybersecurity, and the latest FDA effort is to create draft guidance that expects security throughout the total product life cycle (TPLC).
Another effort is the Protecting and Transforming Cyber Health Care Act of 2022 (PATCH Act of 2022),which, if passed, would revise the existing Federal Food, Drug, and Cosmetic Act.
The FDA guidance establishes six broad expectations on the Secure Product Development Framework (SPDF), which covers all aspects of a product’s life cycle, for the development, release, support, and decommission and satisfy Quality System Regulations (QSR) under 21 CFR Part 820:
- Cybersecurity is a fundamental part of device safety and the QSR
- Security by design
- Transparency
- Security risk management
- Security architecture
- Testing/objective evidence
The FDA draft guidance, under QSR, also declares that verification and validation activities by the medical device manufacturers shall include sufficient testing performed on the Cybersecurity of the system, which validates their inputs and outputs.
Further, the FDA summarizes that cybersecurity controls require testing beyond standard software verification and validation to demonstrate that the device has a good assurance of safety and effectiveness.
The following cybersecurity testing and corresponding objective evidence would be considered as the minimum support for a premarket submission:
Security requirements
- Evidence of their boundary analysis creates a rationale for their boundary assumptions.
- Threat mitigation
- Evidence that all the design input security requirements were implemented successfully
- Evidence of testing their threat models that demonstrates effective risk control measures provided in the system and use case
- Evidence of the adequacy of risk control.
Vulnerability testing – Evidence on the testing of malformed
- Abuse case and unexpected inputs
- Vulnerability chaining
- Closed box testing of known vulnerability scanning
- Software composition analysis of binary executable files
- Static and dynamic code analysis
Penetration testing – Identify and characterize security-related issues that discover security vulnerabilities in the product.
Regular interval cybersecurity testing – It is performed at regular intervals to identify the potential vulnerabilities before exploitation
THE FDA’S ROLE IN MEDICAL DEVICE CYBERSECURITY
Dispelling Myths and Understanding
Download the Fact Sheet (PDF – 175kb)
04/07/2022 Draft Guidance: Cybersecurity in Medical Devices: Quality System Considerations and Content of Premarket Submissions
This draft guidance replaces the 2018 draft version, which emphasizes the importance of understanding that all medical devices are designed securely, enabling new cybersecurity risks to be mitigated throughout the Total Product Life Cycle, and it elaborates the outline of the FDA’s recommendations more clearly for premarket submission to address cybersecurity concerns.
03/08/2022 Cybersecurity Alert: Vulnerabilities identified in medical device software components: PTC Axeda agent and Axeda Desktop Server
The PTC Axeda agent and Axeda Desktop Server are cloud-based technologies that allow people to securely view and operate the same desktop through the Internet. The Axeda agent and its desktop server are owned by the computer software company PTC.
The FDA alerts all medical device users and manufacturers about a cybersecurity vulnerability identified for the Axeda agent and Axeda Desktop Server.
The agent and desktop server of Axeda are used in many medical devices across several medical device manufacturers, and all the versions of the Axeda agent and Axeda Desktop Server are affected.
On the 8th of March, 2022, the Cybersecurity and Infrastructure Security Agency (CISA) published an advisory, ICSA-22-067-01, on these vulnerabilities.
Any successful exploitation of this vulnerability could allow an unauthorized attacker to take complete control of the host operating system, resulting in full system access, remote code execution, reading or changing the configuration, system file access, accessing log information, and other denial condition.
These vulnerabilities may result in changes to the functions of the medical device and impact the availability of the remote support functionality.
As a result, PTC recommends that affected manufacturers:
- To upgrade Axeda agent Version 6.9.2 build 1049 or 6.9.3 build 1051 while running older versions of the Axeda agent.
- Also, to configure the Axeda agent and Axeda Desktop Server to listen only on the local host interface 127.0.0.1.
- Then, Provide a unique password in the AxedaDesktop.ini file for each and every unit.
- Remove the installation file.
- Make sure to delete the ERemoteServer file from the host device.
- Never use ERemoteServer in production.
- When running the Windows operating system, first configure Localhost communications (127.0.0.1) between ERemoteServer and Axeda Builder.
- When running in Windows or Linux, only allow connections to ERemoteServer from trusted hosts and block all others.
- Configure the Axeda agent for the authentication information required to log in to the Axeda Deployment Utility.
So, Cybersecurity is one of the crucial aspects of today’s fast pacing digital world. The threats caused by Cybersecurity, especially on medical devices, are hard to deny. It is important to learn how to defend themselves from them and create a safe environment for the usage of medical devices.
EU MDR and IVDR
In the EU, both the MDR and IVDR Annex I create requirements for mandate consideration of medical device cybersecurity, and the Medical Device Coordination Group (MDCG), in its guidance, explains to the manufacturers of medical devices how to fulfil all the relevant essential requirements regarding Cybersecurity.
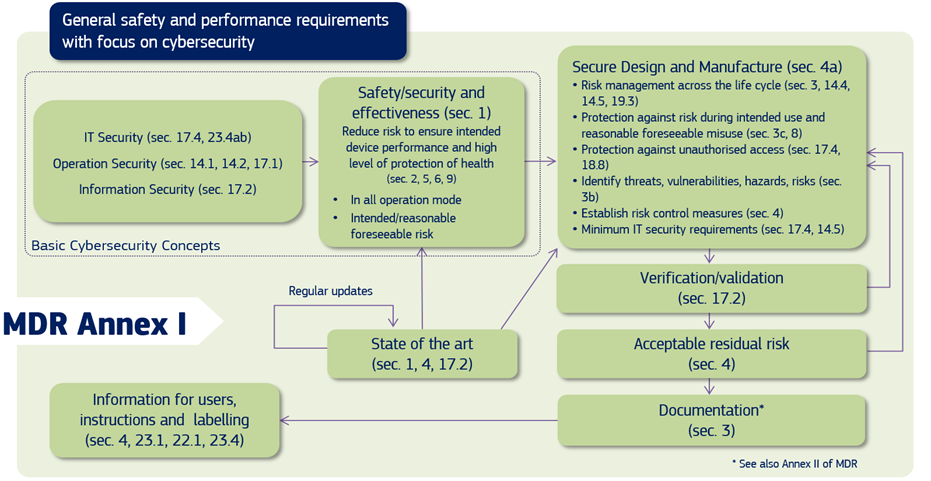
Figure 1: Cybersecurity requirements contained in MDR Annex I
The NIS Directive also provides for legal measures to increase the overall level of Cybersecurity in the EU.
GDPR (General Data Protection Regulation) helps the manufacturers of medical devices in regulating, protecting and processing personal data by the individual, company or organization that relates to the EU.
The EU Cybersecurity Act certifies Cybersecurity for ICT products, services, and processes.
According to the Cybersecurity Act, manufacturers are required to demonstrate state of art in the design, development, and improvement of their medical devices throughout their life cycle.
During that period, the manufacturers must consider the safety, security, and efficacy of the medical devices, and in vitro diagnostic safety mechanism design must be considered early during the manufacturing process.
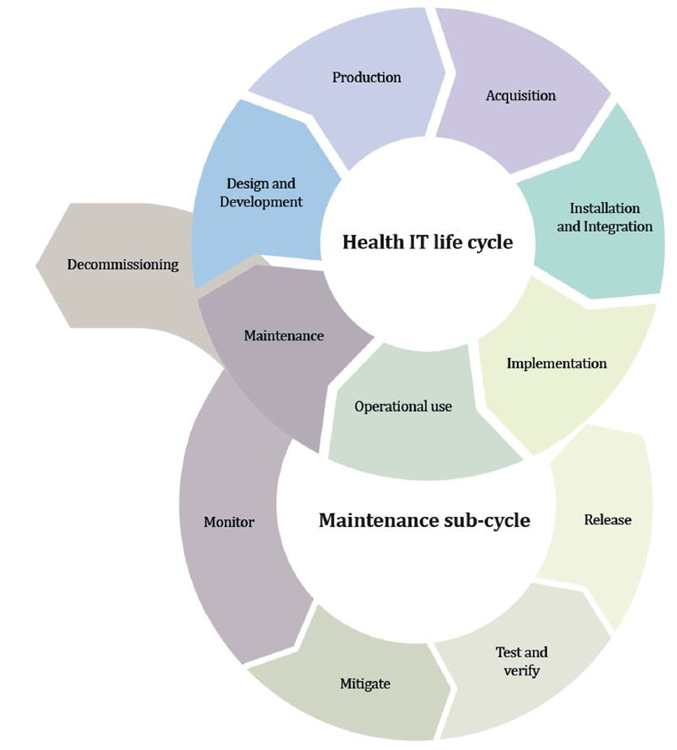
Figure 4: Lifecycle stages
The MDCG has proposed some key philosophies of the staged security concept strategy (“defense in depth strategy”) as follows:
- Security management
- Specification of security requirements
- Security by design
- Secure implementation
- Management of security-related issues
- Security update management
- Security risk management
The list of possible IT security requirements for the operating environment according to MDCG:
- Compliance with national and EU regulations (e.g., GDPR).
- Ensuring appropriate security controls are in place
- Ensuring the physical security of the medical device through security measures
- Ensure control and security of network traffic through proper measures
- Life Cycle Aspects
- Security measures specific to their workstations connected to the medical device.
- Security vulnerabilities related to the device hardware/software and third-party hardware/software used with the medical device.
- During the life of the devices, the manufacturer should implement the process to collect post-market information about the security of the device.
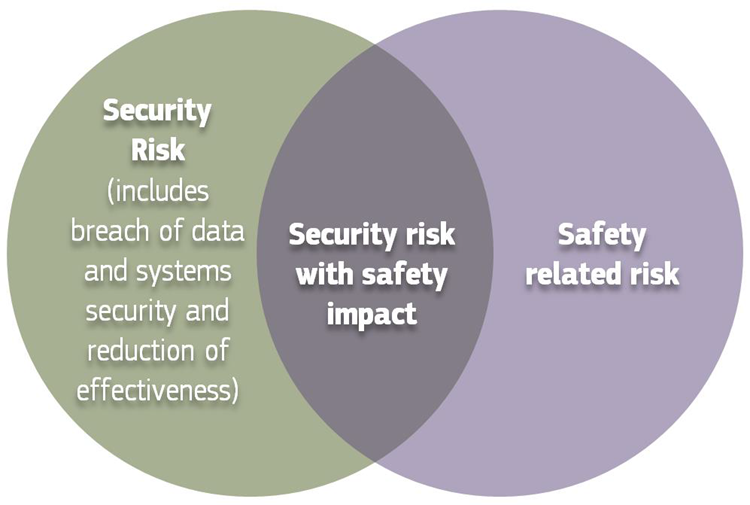
Figure 3: Cybersecurity measures may cause safety impacts
Based on the EU Cybersecurity Act, the manufacturer must provide the following information to the user of the medical device:
Specifications of the operating system
- IT security risk assessment information.
- Provisions for ensuring the integrity of software updates and security patches
- Product installation
- Security configuration options
- Initial configuration guidelines
- Step-by-step instructions for deploying security updates
- Description of the backup and restore functions for data and configuration settings
- Procedures for using all the medical devices in failsafe mode
The manufacturers are required to establish a post-market surveillance (PMS) system and actively keep these PMSs (Post Market Surveillance) up to date. Medical device cybersecurity requirements should be part of this PMS system.
Depending on the class of medical device, a PMS report or PSUR report will be generated, which concludes the analysis of all data from the market.
FAQs
How can we protect heath care from cyber-attacks?
· Vulnerability assessment and required testing
· Training health care providers to protect from any breaches
· Follow the standards of the regulations
Where is Cybersecurity used?
Cybersecurity helps in protecting the Datas, software or hardware connected with the system. This reduces unauthorized access to the data or the system.
What is the PATCH act?
PATCH act helps to meet all the Cybersecurity requirements for the manufacturer to complete FDA regulation standard.
What medical devices can be hacked?
MRI, Pacemakers, Implants, Heart rate monitors, Drug infusion pumps, medical records and other devices connected to the hospital network.
What are the new cybersecurity requirements according to EU MDR?
MDR Annex I explain the risks associated with the interaction between software and medical devices. Manufacturers should follow standard during life cycle, risk management, verification, and validation of the devices.
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.





