Medicine/Drug/OTC/Pharmaceutical Registration in Albania
The Agency for Medicinal Products and Medical Devices (AMPMD) is responsible for overseeing the drug registration process in Albania.
Here is a general outline of the drug registration process:
1. Preparation of Documentation
Collect and prepare all necessary documentation, including but not limited to:
1. 1. Pharmaceutical documentation (composition, manufacturing, and control data)
1. 2. Non-clinical and clinical data
1.3. Information on packaging and labeling
1.4. Summary of product characteristics (SmPC) and patient information leaflet (PIL)
1.5. Quality risk management documentation
2. Application Submission
Submit the completed application and required documents to the AMPMD.
3. Review and Evaluation
The regulatory authority reviews the submitted documentation to assess the safety, efficacy, and quality of the medicinal product. An evaluation committee may be involved in the assessment process.
4. Quality Control Inspection
Conduct a quality control inspection, which may include an inspection of the manufacturing facilities, to ensure compliance with good manufacturing practices (GMP).
5. Clinical Trial Data Review
If applicable, review clinical trial data to support the safety and efficacy of the medicinal product.
6. Labeling and Packaging Review
Evaluate the proposed labeling and packaging to ensure compliance with regulatory requirements.
7. Decision and Approval
Based on the review, the regulatory authority decides whether to approve the drug registration. If approved, a marketing authorization is issued.
8. Post-Market Surveillance
Implement post-market surveillance measures to monitor the safety and effectiveness of the drug once it is on the market.
Documents required for Pharmaceutical Registration in Albania
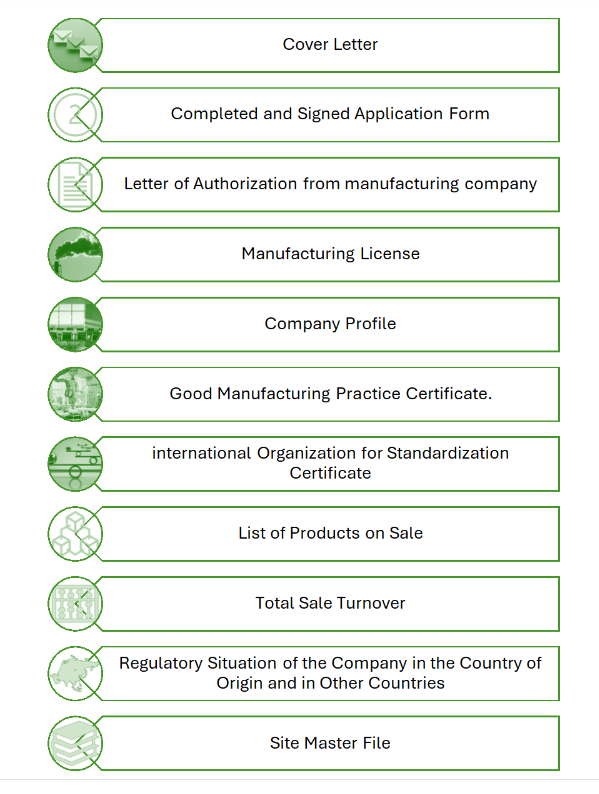
The specific documents required for the registration of medicinal products with the Agency for Medicinal Products and Medical Devices (AMPMD) in Albania can vary based on the type of product, its classification, and the regulatory requirements in place. However, in general, the following documents are commonly requested during the registration process:
1. Application Form: A completed application form provides essential details about the medicinal product, its manufacturer, and other relevant information.
2. Pharmaceutical Documentation: Information on the composition of the medicinal product, including active ingredients and excipients. Manufacturing and control data, including details about the production process, quality control methods, and specifications.
3. Non-Clinical Data: Non-clinical study reports that demonstrate the safety and pharmacological properties of the medicinal product.
4. Clinical Data: Clinical trial data (if applicable) demonstrating the safety, efficacy, and quality of the medicinal product. Summary of clinical trial results.
5. Quality Risk Management Documentation: Documentation related to quality risk management, which assesses potential risks and outlines strategies for mitigating those risks.
6. Labeling and Packaging Information: Proposed labeling and packaging information, including the Summary of Product Characteristics (SmPC) and Patient Information Leaflet (PIL).
7. Good Manufacturing Practices (GMP) Certificate: A GMP certificate for the manufacturing site(s) of the medicinal product.
8. Declaration of Conformity: A declaration confirming that the medicinal product complies with relevant regulatory requirements.
9. Power of Attorney: If applicable, a power of attorney or authorization allowing a representative to act on behalf of the applicant.
10. Fees: Payment of the required registration fees.
Pre-Market Afghanistan Drug Registration Procedure
In the ever-changing healthcare landscape of Afghanistan, it is critical to effectively regulate medical goods and services. This paper explores the complex pre-marketing processes, including planning and policy, licensing, marketing permits, supply, price, and the vital oversight and management of clinical trials.
1. Policy & Plan
Planning and Coordination is the first step towards a strong regulatory system. The Ministry of Public Health in Afghanistan assumes a leading role in coordinating the efforts of many parties participating in the pre-marketing phase. Manufacturers, scientists, medical experts, and governmental organizations fall under this category. The foundation for efficient cooperation and communication is laid by strategic planning.
The success of pre-marketing operations depends heavily on clear and efficient communication. It is necessary to keep stakeholders updated on guidelines, regulations, and policy changes. Clear and frequent communication channels are set up to provide manufacturers, distributors, and other pertinent parties with information.
Information is a key component of regulatory decision-making, according to information management and evaluation (M&E). Strong data management systems make sure that data is gathered, examined, and assessed about clinical trials, healthcare goods, and market dynamics. Mechanisms for routine monitoring and assessment are in place to determine where improvements might be made and to gauge the success of current policies.
The foundation of healthcare regulation is the drafting and revision of legislative documents. Lawmakers in Afghanistan create and amend laws on a regular basis to meet changing needs and conform to global norms. Carefully constructed legislative documents handle licensing, permits, cost, and clinical study regulation.
2. Licensing
This essential pre-marketing step makes sure that only safe and efficient medical supplies reach consumers. A rigorous vetting process is required of businesses looking to sell their goods in Afghanistan. Complete information provided during the licensing application is used by the regulatory body to evaluate the product’s quality, safety, and efficacy.
3. Permission to Price and Supply the Market (Valuation)
Supply Chain Oversight: Businesses need permits to sell their goods to keep a safe and effective supply chain. By ensuring that products are supplied and distributed in accordance with regulatory criteria, this reduces the possibility that inferior or counterfeit goods may enter the market.
Pricing and Valuation: Accessible healthcare is facilitated by the establishment of reasonable pricing structures and the appraisal of healthcare items. To establish fair prices, regulatory agencies collaborate with manufacturers, considering aspects including quality, production costs, and market conditions. The goal of this procedure is to achieve a balance between sustainability and affordability.
4. Control and Regulation of Clinical Research
Clinical research is essential to expanding our understanding of medicine and guaranteeing the security and effectiveness of medical treatments.
To safeguard participants and maintain ethical standards, clinical research are tightly regulated and controlled by Afghan regulatory organizations. This method includes constant monitoring, informed consent processes, and a close examination of study protocols.
Post-Market Afghanistan Drug Registration Procedure
Beyond the original drug registration procedure, there is a continuous commitment to ensuring the safety, efficacy, and quality of pharmaceuticals.
1. Examining and Implementing Laws and Rules
Supervisory Authority: Afghanistan passionately believes that post-market inspections are essential to upholding pharmaceutical standards. To make sure that manufacturing sites, distribution networks, and retail locations are adhering to laws and regulations, regulatory bodies regularly audit them. The goal of this thorough inspection is to find and address any deviations from accepted norms.
Cooperation with Stakeholders: Afghan regulatory agencies work closely with pharmaceutical companies, medical practitioners, and other stakeholders to improve post-market surveillance. This cooperative strategy encourages a shared accountability for maintaining pharmaceutical standards, with continual education and communication to address issues and encourage adherence.
2. Pharmacovigilance, or Market Surveillance
Pharmacovigilance Framework: An essential part of Afghanistan’s post-market processes is market surveillance via pharmacovigilance. Data on the safety of pharmaceutical products are systematically collected, analysed, and interpreted as part of the pharmacovigilance framework. This continuous procedure aids in identifying and evaluating side effects or any other issues relating to drugs.
Adverse Event Reporting: It is crucial for patients, healthcare providers, and pharmaceutical companies to report adverse occurrences connected to their products. There is a strong reporting mechanism in place that makes it possible to promptly submit data on unanticipated side effects or other safety issues. To decide what steps to take next, regulatory bodies thoroughly look at these reports.
Risk management and signal detection: One aspect of pharmacovigilance work is the ongoing observation of signals that can point to safety issues. Regulatory agencies strive to identify new dangers and proactively address them. This could entail changing the product’s label, releasing safety alerts, or, in the worst situations, taking the medication off the market.
Documents Required for Product Registration in Afghanistan
1. Official introduction-letter and addresses of the company issued by the company itself.
2. License of producing medicine and medical equipment and certification of three authorized organizations of the origin country and that of WHO.
3. Special license for producing specific items from MoPH and certifications from high authorities of the origin country and that of WHO.
4. License of exportation.
5. Documents for the usage of the products inside the origin country.
6. Quality control documents of the company and also quality control documents from the central laboratory and ministry of MoPH of the origin country.
7. Specific documents of the medicine such as: list of formulation including standard formulation of the medicine, list of prices including reasonable prices, packing list with the specification about packing material, and method of packing. Procedure for controlling the production process, specification of its contents, documents of analysis, method of analysis, reliability of the method of analysis certification of the contents and pharmacological report of the medicine.
8. Original stamp of the company on the relevant documents.
9. Certification of the ministry of MoPH and stamp of chambers of commerce of the origin country and issuance of them through trading department of the embassy of Afghanistan located on the producing country.
10. Analysis the sample of the medicine in the general medicine analyzing laboratories of Afghanistan and registering the mentioned products.
Registration Requirements for Product Registration in Afghanistan
Fees for Afghanistan Drug Registration
AFN (50,000)
License Validity for Afghanistan Drug Registration
3 years.
Local Authorized Representative
Yes, a local Authorized Representative is required before you place your product on the market.
A medical device’s intended use and inherent risks must be considered when determining its MDR classification. Class I devices pose less risk to patients and end users, as under the previous MDD. The new Regulation EU MDR 2017/745 has added extended rules, leading some devices to fall under Class IIa, IIb, or even III.
This document is intended to guide Class I medical device manufacturers (excluding custom-made devices) that sell products bearing their name or trademark on the Union market to comply with MDR requirements. This guidance should also apply when an importer, distributor, or any other legal entity takes on the obligations owed by manufacturers.
The class I manufacturer must also hire a Notified Body (NB) to perform a conformity evaluation if medical devices are to be provided sterile, have measuring functions, or are reusable surgical instruments. For the NB to be qualified to conduct such a conformity assessment, the specific medical device in question must come within the purview of the Notified Body’s designation.
Large and mid-sized medical device manufacturers must also properly designate at least one individual in charge of regulatory compliance, but micro and small companies can function better by having a regulatory expert.
It is also important to designate an authorised representative residing within the EU if the manufacturer is registered outside of the EU. A foreign manufacturer must specifically issue a mandate outlining the representative’s duties and authority. It is also crucial to note that not all responsibilities associated with the medical equipment can be assigned.
The manufacturer must adequately preserve all the records required to verify compliance with the relevant requirements so they may be made available to the regulatory body upon request.
Steps to placing Class I medical devices on the market:
Manufacturers must ensure compliance with each of the following requirements if they plan to commercialise Class I medical devices. Please be aware that some of the outlined requirements are interconnected and can be completed in a different sequence than that which is shown.
The manufacturer will carry out a gap analysis for Class I devices that have already been released onto the market in compliance with the MDD to ensure that all of the below-listed requirements were satisfied at the time the MDR was applied.
The whole procedure includes a set of mandatory steps, including:
Step 0: Integrate MDR in the Quality Management System (QMS)
The manufacturer’s QMS should be appropriately linked with the standards outlined in the MDR. To ensure compliance with the following requirements, this will enable the correct assessment or decision to be taken and the appropriate documented evidence to be produced.
Step 1: Confirm product as a medical device
The intended purpose and the product’s mode of action are reviewed as per Article 2(1) of the MDR to ensure that the product qualifies as a medical device. If a product is assigned multiple intended purposes, it will qualify as a medical device only if all the intended purposes are covered in Article 2(1).
In the case of products identified as more than one category, the relevant legislation requirements will have to be followed. Despite not being medical devices in and of themselves, accessories to medical devices are subject to MDR regulations and qualify as devices under the MDR definition.
However, the MDR does not include accessories to devices covered by it as per Annex XVI of the MDR.
Step 2: Confirm product as a Class I medical device
The product has to be confirmed if it can be classified as a class I device as per Annex VIII of the MDR. Products previously classified as class I under MDD should be reviewed according to the classification rules of MDR to confirm if reclassification is required.
The MDD guideline cannot be used for devices that were moved from Class I to higher risk classes by the adoption of the MDR. The intended use of the device and any inherent risks related to the period of use, the part of the body, whether it is active or not, and whether it is invasive or non-invasive will determine how the classification standards are applied.
The classification rule with the highest class should be applied if the device in question falls under the purview of more than one classification rule due to its attributes.
Step 3: Procedures before placing on the market
a) Meet the General Safety and Performance Requirements (GSPR)
The devices will adhere to the general safety and performance standards outlined in Annex I of the MDR, considering the intended purposes that their manufacturers had provided.
The Risk management system is a continuous iterative process throughout the entire lifecycle of the product, established by the manufacturer that will enable the identification and analysis of the risks related to each device, the estimation and assessment of the risks associated with those risks, the elimination or control of residual risks, and the evaluation of the adopted measures based on the data gathered from the PMS system.
When a harmonised standard is in place, but a manufacturer chooses to use another reference, implementing that reference should, at the very least, ensure the same level of performance and safety.
Compliance with the relevant harmonised standards will give rise to a presumption that the MDR’s requirements, or portions of them, are also complied with.
If there are available standard specifications, the manufacturer must adhere to them unless they can adequately demonstrate that they have chosen a solution at least as effective and safe. The clinical evaluation processes, risk management, and PMS must be interdependent and updated regularly.
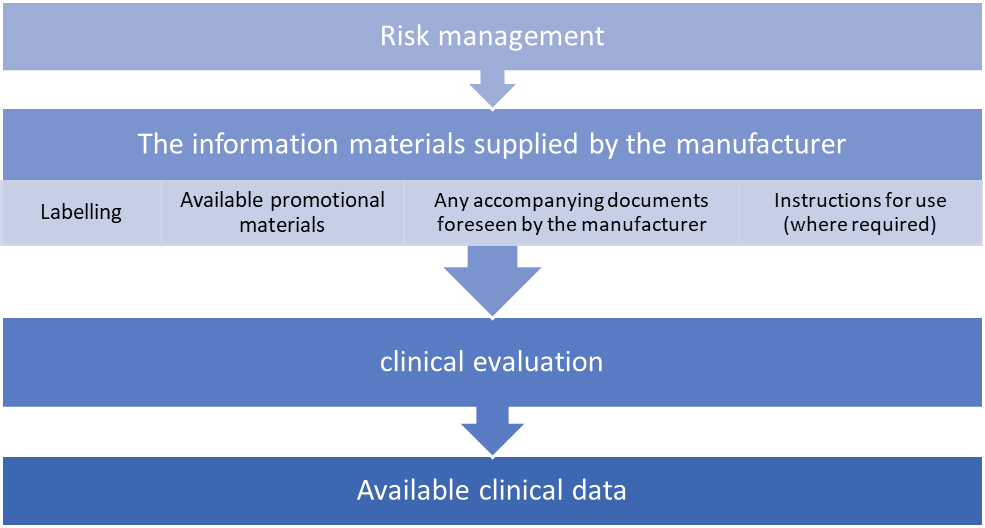
b) Conduct clinical evaluation
As part of the MDR’s technical documentation requirements, all devices—regardless of risk classification—require a clinical evaluation. The level of clinical evidence required to show compliance with the pertinent general safety and performance requirements listed in Annex I, which is obtained by considering the characteristics of the device and its intended purpose, shall be specified, and justified by the manufacturer.
Manufacturers must prepare, carry out, and record a clinical evaluation in compliance with Article 61 and Part A of Annex XIV to accomplish this.
Conformity to Annex I requirements can only be assumed when the following items are aligned with each other.
Consideration of available alternative treatment options, the incorporation of clinical data and the acceptability of the benefit-risk ratio are required for carrying out a clinical evaluation.
Additional clinical data will be acquired or developed by clinical investigations if the currently available clinical data are insufficient to establish compliance with the MDR.
If the clinical data currently available for a device that is currently certified under Directive 93/42/EC is insufficient to show compliance with MDR, then post-market clinical follow-up studies of the device may be used to gather more clinical data.
Even data from the general post-market follow-up may occasionally be enough to close the difference.
a) Prepare technical documentation
The manufacturer is responsible for creating and maintaining the technical documentation to prove that their products adhere to the MDR’s technical specifications. Under Annex II and III, this technical documentation must be prepared before the EU declaration of compliance is drawn.
The manufacturer will develop and provide the technical documentation and, if applicable, it’s summary in a way that is unambiguous, clear, well-organised, and easy to search.
The manufacturer shall provide the CA, the authorised representative (where applicable), and NB with access to the technical documents (when applicable).
After reviewing the general safety and performance standards, as well as the pertinent technical provisions of the MDR, the technical documentation will be developed.
b) Request Notified Body involvement
Class I devices do not require the engagement of an NB for MDR compliance. But the manufacturer must follow the guidelines outlined in Chapters I and III of Annex IX or Part A of Annex XI of MDR when the product is a sterile equipment, measuring instrument, or reusable surgical tools.
For the relevant codes and corresponding types of devices as established by Regulation (EU) 2017/2185, manufacturers may select any NB designated in accordance with the MDR.
c) Prepare Instructions for Use and Labelling
Any safety and performance information required for a device’s safe usage and identification of the device should be provided along with the device by the manufacturer and/or the authorised representative, considering the possible users’ training and knowledge.
This data is included on the label, in the device packing, and in the instructions for use. Class I devices do not need instructions for use if they may be operated effectively and securely without them, deviating from the general rule.
Since Class Ir devices will need instructions for reprocessing (cleaning and sterilisation), an exception is most likely proposed.
Labelling and instructions for use must be written in accordance with national language regulations. There will be versions of the labelling and IFU in the technical documentation (in each pertinent national language).
The requirements regarding the information to be supplied with the device will be found in Annex I, Chapter III (23).
Step 4: Check compliance with general obligations for manufacturers
The manufacturer shall ensure that the general obligations for manufacturers outlined in Article 10 are met before releasing a device for sale.
Implementing a suitable QMS that will most effectively assure compliance with the MDR, such as through an internal audit, will receive special consideration. The QMS shall be documented, applied, maintained, updated regularly, and developed continuously, and it will at least include the following elements:
A strategy for regulatory compliance
Identification of applicable general safety and performance requirements and exploration of options to address those requirements
Responsibility of the management
Resource management, including selection and control of suppliers and sub-contractors
Risk management
Clinical evaluation, including post-market clinical follow-up (PMCF)
Product realisation, including planning, design, development, production and service provision
Verification of the UDI assignments
Setting up, implementation and maintenance of a post-market surveillance system
Handling communication with competent authorities, notified bodies, other economic operators, customers and/or other stakeholders
Processes for reporting serious incidents and field safety corrective actions in the context of vigilance
Management of corrective and preventive actions and verification of their effectiveness
Processes for monitoring and measurement of output, data analysis and product improvement.
According to applicable Union and national law, either natural or legal persons may seek compensation for harm brought on by a defective equipment.
In order to protect themselves against potential responsibility under Directive 85/374/EEC, manufacturers must take precautions that are commensurate to the risk class, type of device, and size of the business. These steps must not compromise further protective measures under national legislation.
Step 5: Draw-up the EU Declaration of Conformity
The process by which the manufacturer, who complies with the standards established by Article 52(7), certifies that the devices in question comply with the requirements of the MDR that apply to them is referred to in Article 19 as the EU declaration of conformity.
The CA will have access to the declaration of compliance, which must include, at a minimum, all of the data referred to in Annex IV. The manufacturer will regularly update the EU declaration of conformity and translate it into the official language(s) required by the Member States where the product is sold.
Suppose a device is subject to additional Union laws in addition to the MDR that also call for an EU declaration of conformity. In that case, the manufacturers will create a single EU declaration of conformity that refers to all applicable Union laws.
By creating the EU declaration of conformity, the manufacturer accepts liability for the device’s regulatory compliance with all relevant Union legislation.
For class Ir, Im, and Is devices, the manufacturer must obtain an EC certificate from NB per Annex IX, Chapters I and III, or Annex XI, Part A before applying a CE mark.
Step 6: Affix the CE marking
All Class I medical devices on the market must display the CE conformity label, which must be visible, readable, and permanent. It may be applied to the item or its sterile packaging. The CE marking must be applied to the package in cases where such affixing is impossible or unwarranted due to the nature of the device. The CE certification must be visible on all sales packaging and the instructions for use.
Placing the CE mark on a medical device signifies that it complies with all applicable safety and performance standards and is approved for marketing in the EU. The CE marking will be accompanied by the identification number of the relevant NB in the case of Class I medical devices placed on the market in a sterile condition, devices with measurement functions, and/or reusable surgical tools.
Affixing marks that could lead third parties astray about the significance of the CE marking is banned. Other extra markings may be applied to the product, the packaging, or the usage instructions, but they must not obscure or obstruct the CE marking.
The CE marking format will follow Annex V requirements. The minimum dimensions required for the CE mark may not apply to very small devices.
Step 7: Registration of devices and manufacturers in Eudamed
A streamlined approach might be used for medical devices that were previously put on the market per the Directives, provided that the manufacturer evaluates the gap and ensures that all regulations are properly followed.
A Class I medical device manufacturer must register the product with Eudamed before putting it on the market. Suppose the information listed in Section 1 of Part A of Annex VI has not already been registered in accordance with Article 31. In that case, the manufacturer must submit it to the electronic system in order to register the device. The information referred to in Section 1 of Part A of Annex VI will be given to that electronic system before applying to the NB in situations where the conformity assessment method necessitates the engagement of an NB in accordance with Article 52.
Following the CA’s validation of the manufacturer’s data in Eudamed, the manufacturer will receive an SRN from the aforementioned electronic system. To fulfil its duties under Article 29, the manufacturer will use the SRN when submitting an application to an NB for a conformity assessment and to gain access to Eudamed.
The registration of a device in Eudamed by the manufacturer includes:
Assigning a UDI-DI (with a Basic UDI-DI) as described in Part C of Annex VI to the device in accordance with the issuing entity’s policies mentioned in Article 27(2) and adding the UDI-DI (with a Basic UDI-DI) to the UDI database along with the other core data elements pertaining to that device mentioned in Part B of Annex VI.
Entering the data referred to in Section 2 of Part A of Annex VI, excluding Section 2.2 thereof, or, if previously provided, validating the data, and then keeping the data current up to date.
This document summarises the information presented above and outlines how Class I medical devices should be introduced to the EU market. The guidance’s scope also includes reusable surgical instruments (Class Ir), sterile medical devices (Class Is), and devices with measurement capabilities. These medical devices all need a Notified Body to be involved in pre-marketing procedures.
Technical documentation should contain details of the medical device in a clear, organized, readily searchable and unambiguous manner.
It should be provided with all medical devices irrespective of the device class and to be kept updated throughout the product life cycle. Technical documentation is to be prepared according to the requirements given in Annex II of EU MDR 2017/745.
Requirements of Technical Documentation as per Annex II
Device description and specification, including variants and accessories
Information to be supplied by the manufacturer
Design and Manufacturing information
General safety and performance requirements
Benefit-risk analysis and risk management
Product verification and validation
Device Description and Specification, including variants and accessories
This section mentions that technical documentation must include all basic device details such as trade name, general description, basic UDI-DI, principles of operations, classification details, accessories description, key functional elements, technical specification etc. There must also be a proper reference to previous or similar medical devices.
Information to be supplied by the Manufacturer
This section mentions that manufacturer must include a complete set of labelling for the device and its packaging. The manufacturer must also ensure the language requirements of the same according to the each member states’ language requirements.
Design and Manufacturing Information
This section mentions that the technical documentation shall include complete information on processes carried out by the manufacturer during the design stage, manufacturing, validation, monitoring and final product testing of the device.
Identification details of all sites and people involved in the manufacturing process must also be made available.
General Safety and Performance Requirements
This section mentions that the technical documentation must demonstrate conformity to general safety and performance requirements set out in Annex I of the MDR.
This shall include details such as an explanation for safety and performance requirements that the device satisfies and an explanation for requirements that it does not meet, methods used to demonstrate conformity, harmonized standards, Common specifications, and the identity information of the control documents.
Benefit-Risk Analysis and Risk Management
This section mentions that the technical documentation shall include the benefit-risk analysis, solutions adopted, and risk management results as per Annex I of the MDR.
Some significant points from the Risk management section of Annex I are
The risk management plan must be available for each device
Should be able to identify foreseeable hazards associated with each device
should be able to estimate and evaluate the risks
should have plans to eliminate/control the risks
should be able to assess the impact of risk at various stages of the device’s lifecycle and thereby estimating overall risk, benefit-risk ratio, and risk acceptability
Section 1 to 8 of Annex I gives detailed information on general safety measures and other risk management requirements.
Product Verification and Validation
This section mentions that the technical documentation shall contain results and critical analyses of all verification and validation tests/studies.
Some significant Tests/studies discussed in the section are
Biocompatibility
Electrical Safety
Electromagnetic compatibility
Software verification and validation
Stability
Performance and Safety
Physical, chemical, and microbiological characteristics
sterility
A clinical evaluation report and a PMCF (Post-market clinical follow-up) plan and evaluation report must be available. Else, a justification for why PMCF is not applicable must be made available.
Additional information required in specific cases is given in detail in Section 6.2 of Annex II of the EU MDR.
Post-Market Surveillance
Annex III of the MDR majorly mentions that the post-market surveillance plan must address the method of collection and utilization of available data (Such as information on serious incidents and their corrective actions, data on any undesirable side- effects and so on) and includes a list of details a post-market surveillance plan must include in the plan.
There is also a reference to PSUR (Periodic safety update report) and post-market surveillance report mentioned in articles 86 & 85, respectively.
Significant Changes to be noted during the transition from MDD to MDR
Device description to include UDI, UDI-DI or any such number for traceability reasons
Reference requirements to previous or similar medical devices
Under labelling requirements in the MDR, it is explicitly talks about the labelling of devices and their packaging
Language requirements for labelling
As per the MDD, only Class III devices required an explanation for design stages and procedures. The same is now updated to all classes of medical devices in MDR
Identification requirements of all manufacturing sites, design including suppliers and contractors are required now
General safety and performance requirements are updated versions of Essential requirements from the MDD
The benefit-risk analysis is explicitly mentioned as part of risk management requirements in the MDR
Product verification and validation requirements
A detailed explanation of pre-clinical and evaluation and Clinical Evaluation requirements are provided in the MDR
The requirements of the Clinical Evaluation plan and report, Post-market clinical follow-up plan and report are specially mentioned in the MDR
Additional information for special case devices (e.g., combination device, sterile device, device with measuring function etc.) is mentioned in detail in the MDR
FAQs:
What are Common specifications?
Common Specifications are a set of technical and/or clinical requirements, other than a standard, that provides a means of complying with the legal obligations applicable to a device, process or system.
What is the UDI?
The UDI is a series of numeric or alphanumeric characters that is created through a globally accepted device identification and coding standard. It allows the unambiguous identification of a specific medical device on the market. The UDI is comprised of the UDI-DI and UDI-PI. The unique identifier may include information on the lot or serial number and be able to be applied anywhere in the world.
What is the Basic UDI-DI?
Basic UDI-DI is intended to identify and connect devices with the same intended purpose, risk class and essential design and manufacturing characteristics. It is independent/separate from the packaging/labelling of the device and it does not appear on any trade item
Is Post-market surveillance (PMS) applicable for only higher risk classes?
PMS is required for all device classes. A PMS plan and report to be maintained and kept updated by the manufacturer throughout the device life cycle.
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.
“Professional use” Test Kit – Procedure for registration in UK
Novel Corona Virus Disease 2019 (COVID-19) is a different variety of disease that has never been seen in humans before, impacting being a pandemic, and affecting the world since the year 2020.
One of the most difficult aspects of coronavirus is the ambiguity of not knowing who has been infected or whether it is safe to resume normal activities. Testing of high quality might assist provide assurance.
However, no good thing in the world has ever been without challenges. Accuracy and reliability of the tests, getting the right supply of people, lab space, equipment and chemicals, and logistics has proven to be the major hurdles in the testing procedures.
Nevertheless, governments across the globe are striving to cope up with these challenges for bettering the lives of the masses.
Out of a variety of Tests available in the UK Market, a Professional use test kit, as the name suggests, is intended to be used by the professionals.
By professionals, it means the kits to be used by personnel who have obtained specialized training and education in processes involving in-vitro diagnostic medical equipment, such as laboratory personnel, trained clinicians, and qualified healthcare professionals.
This kit must be UKCA, or CE marked, and must be categorized as general IVD as per the amended UK MDR 2002. If the manufacturer of a professional use COVID-19 test expects the test kit to be used in an assisted or supervised testing condition, the IFU should state so, and there should be documentation of performance statistics to back up this claim.
Talking about the test kit regulations as per UK’s Ministry of Health – Medicines and Health products Regulatory Agency (MHRA), no antigen or molecular detection COVID-19 (SARS-CoV-2) test may be placed on the UK market without first being verified against minimal performance standards through a Coronavirus Test Device Approvals (CTDA) desktop evaluation, according to regulation 34A of the Medical Devices Regulations 2002.
Manufacturers or distributors supplying COVID-19 test kits must apply to the Department of Health and Social Care (DHSC) for approval and their product must meet The Medical Devices (Coronavirus Test Device Approvals) Regulations 2021.
The MHRA will not accept registration applications for covid test devices until they have obtained CTDA approval or have been placed on the Temporary Protocol list.
If the manufacturer believe equipment is exempt from the CTDA Regulations 2021 and want to register it with the MHRA, manufacturer will most likely be contacted to explain the exemption applicable in the regulations before the MHRA registration application is accepted.
Steps for applying to CTDA approval
Step 1: Submitting the application
Make sure to provide the below data properly:
Manufacturer and product information
Regulatory status
Product performance
Biosafety
Supplementary documents (for example: current version of the instructions for use, biosafety documents, evidence of performance characteristic)
Step 2: Desktop Review
It is the assessment of the evidence submitted by a supplier against a minimum required data set.
This stage prohibits products to reach the market that do not meet the expected criteria from the CTDA Regulations 2021.
The main areas of assessment are below:
Manufacturer and test information
Regulatory status
Intended use case
Product performance
Biosafety
(Note: If you require a complete checklist of the above areas of assessment, mail to [email protected])
DHSC generally produces the results within 20 working days, but can take longer depending on the availability, and applications volume. If DHSC contacts for more information, it must be responded in 20 working days, else the application might be rejected.
For the approval, it is mandatory to pay the fees, depending on the size of the company applying for validation. The price for desktop review is £6,200 if the company has no more than 250 individuals in total. Otherwise, the cost is £14,000.
DHSC only publishes details of tests that have passed, showing:
Indicative performance category
Name of the test
Name of the manufacturer and business address
Name and business address of person who made application (if different to the manufacturer)
Date and version for instructions of use
Type of test
Date of approval
CE certification number
Country of manufacture
Sample type
If the application is unsuccessful, a request to DHSC for reconsidering its decision can be made using the web form in the service.