This article focuses on the distinctions between the MDD and MDR, considering the terminologies and real-world examples of substance-based devices and combinations of medical and medicinal products.
The guidance distinguished the following three concepts in MDR.
1. Article 2(1) MDR, first paragraph refers to the ‘Specific medical purpose’ specified by the manufacturer as
diagnosis, prevention, monitoring, prediction, prognosis, treatment of disease
diagnosis, treatment, monitoring, alleviation of, or compensation for, disability or injury
the investigation, modification, or replacement of the anatomy or a physiological or pathological process or state
2. Article 2(1) MDR, second paragraph refers to ‘Principal intended action’, which describes the manufacturer’s labelling and claims based on state-of-the-art scientific data regarding the principal mode of action, on a case-by-case basis like
the medical devices for the control or support of conception
products specially intended for the cleaning, disinfection, or sterilization of devices
3. Article 1(6)(b) MDR refers to the ‘Principal mode of action’ by which the product meets its principal intended action, i.e., pharmacological, immunological, metabolic, physical, or other factors.
Substances-based medical devices
A medical device that uses substances as its base contains ingredients approved for use in medical devices and does not carry out its primary intended activity by pharmacological, metabolic, or immunological mechanisms.
Those devices are used equivalently as a medicinal product, for example, ingested or applied to the skin.
In addition to outlining broad guidelines for substance-based medical devices, the guidance offers an explanation and examples for the substances-based devices that fall under two rules according to MDR Annex VIII.
Rule 3 talks about the medical devices containing a chemical or combination of substances utilized in vitro in direct touch with human cells, tissues, organs, or human embryos before implantation or administration
For example
IVF or ART (Assisted Reproductive Technology) products without principal pharmacological/metabolic action (substances or mixture of substances)
IVF cell media without human albumin
Solutions for the transport of organs for transplantation (that do not achieve their principal intended action via pharmacological, immunological, or metabolic means),
Rule 21 talks about the medical devices made of substances or mixtures of substances that are meant to be injected into or applied to the skin
For example
Vaginal lubricants/moisturizing gel
Salt water used for nose or throat sprays
Skin treatment formulations
Eye drops for lubrication
Ear drop
Oral administration of medical devices to treat obesity
Medical Device and Medicinal Products combination
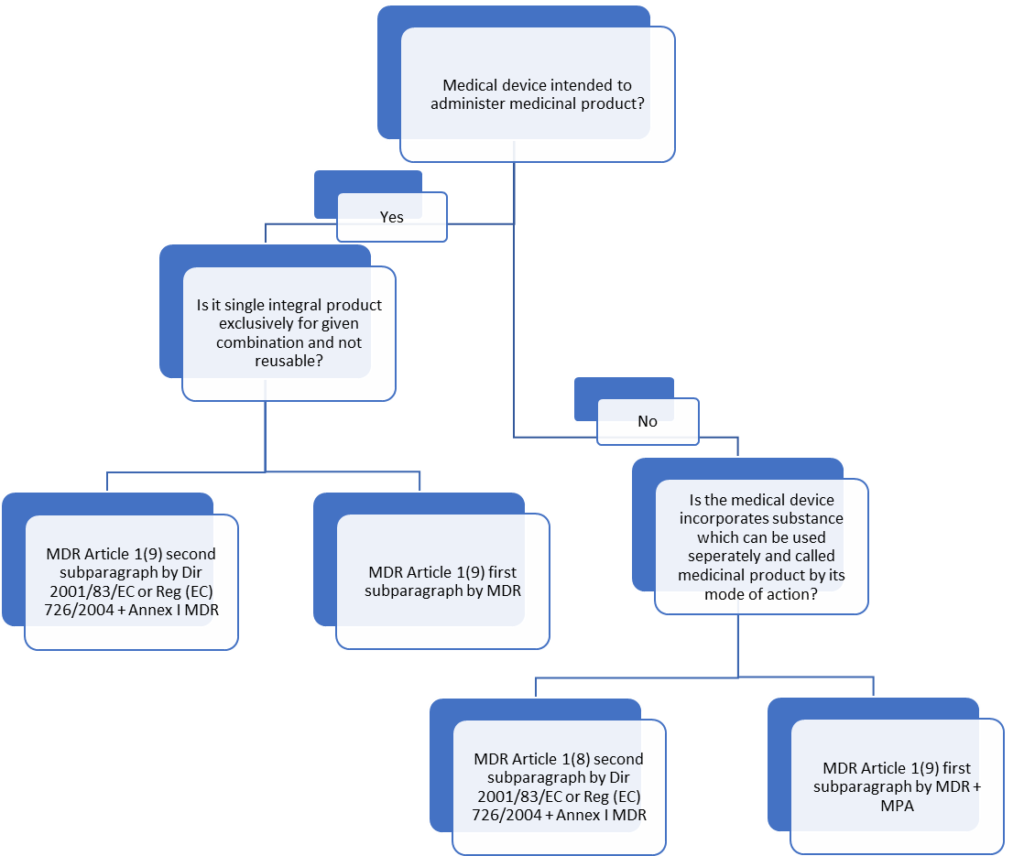
The MDR Article 1(8) and Article 1(9) give a helpful flow chart (Fig 1) for medical devices that are intended for use with a medicinal product in a combination product.
Flowchart to find the regulatory requirement of the combination product
What is meant by “integral” is the key concept in MDCG 2022-5 SECTION 4:
Article 1(8), First Paragraph of MDR, uses the phrase “integral part” to describe components of devices that would be classified as therapeutic items if used independently.
In the second paragraph of Article 1(8) MDR, the term “integral products” refers to devices that incorporate a material that, if taken separately, would be considered a medicinal product.
When a product is on the market, it must have at least two separate components, one of which is a device that, when connected (for example, physically or chemically), functions as a single unit and comes under integral product.
As an exceptional case, medical devices co-packed with a medicinal product or devices included in the information provided with the medicinal product are not considered integral products if the pertinent combination occurs at the time of administration.
Devices used to deliver a medicinal product, and the respective medicinal product forms a “single integral product” are mentioned in Article 1(9), second paragraph MDR.
A single integral product comprises at least two constituent elements, one of which is a device and the other is a pharmaceutical, mixed in a way that prevents their intended separation before use.
If the substance accomplishes the primary intended action of the integral product, the entire product is governed in accordance with Directive 2001/83/EC or Regulation (EC) No 726/2004 as a medicinal product.
If the medical device accomplishes the primary intended function, the complete product is governed by the MDR as a medical device containing a medicinal substance with an additional function.
Devices for administering medications, where the medication is provided separately, are not integral products.
MDCG 2022-5 provides clear definitions with examples for the following:
Combinations of medical devices and medicines are regulated as medicinal products.
The MDR states that Directive 2001/83/EC or Regulation (EC) No. 726/2004 governs the entire product but that the relevant general safety and performance standards of Annex I MDR shall apply as far as the safety- and performance-related device characteristics are concerned.
If the device has a CE mark, the conformity assessment results must be included in the marketing authorization dossier, including the variation dossier.
If the dossier does not contain this information, however, and if a notified body would be required to conduct a conformity assessment of the device if used separately, an opinion on the conformity of the device part with the general safety and performance requirements listed in Annex I MDR must be provided. For further guidance, refer to EMA (European Medicines Agency).
For example
Pre-charged nebulizers with the specific medicinal products
Pre-filled syringes with the specific medicinal product
Devices designed to administer a medicinal product within the meaning of the MPD fall under this category, even when the device and the medicinal substance are not integrated.
For example
Jet injectors
Port systems
Implantable infusion pumps
Drug delivery pumps
Spacers intended for meters dose inhalers
Medical devices with an ancillary medicinal product built in as a component
The MDR outlines the situation in which medical devices contain a substance that, if used alone, would be considered a medicinal product under Article 1 of the MPD, including a medicinal product made from human blood or human plasma, with an action unrelated to the device’s intended purpose which includes herbal medicinal products too.
For example
Bone void fillers containing growth factors
Condoms coated with spermicides
Liquid wound dressing containing the anti-microbial agent
Catheters coated with heparin or an anti-microbial agent
Medical devices that include herbal products
Clove Oil (Caryophylli aetheroleum) – has antiseptic, analgesic, and sedative properties.
Thymus vulgaris (Thyme) – disinfectant, antiseptic and expectorant properties
Medical devices that include human blood or its derivative
Culture media used in IVF containing human albumin solution
Haemostatic agent/matrix containing human thrombin
Annex VIII, Rule 14 states that ‘All devices incorporating, as an integral part of a substance which, if used separately, can be considered to be a medicinal product, as it is defined in point 2 of Article 1 of Directive 2001/83/EC, including medicinal product which derived from human blood or human plasma, as defined in point 10 of Article 1 of that Directive 2001/83/EC, and that has an action ancillary to that of the devices, are classified as class III’.
Furthermore, Annex IX section 5.2(a) requires that ‘Where a device incorporates, as an integral part of a substance which, if used separately, may be considered to be a medicinal product within the meaning of the point 2 of Article 1 of Directive 2001/83/EC, including a medicinal product that derived from human blood or human plasma and that has an action ancillary to that device, the quality, safety and usefulness of substance shall be verified by analogy with the specific methods in Annex I to Directive 2001/83/EC’.
FAQs
What is EMA?
The European Medicines Agency (EMA) is an agency of the European Union which regulates the evaluation and supervision of medicinal products. Its main responsibility is to protect and promote public and animal health.
What is a combination product?
Combination products are two or more components, such as a drug and a device, a drug and a biological, a biologic and a device, or a drug and a device and a biological, must be integrated, mixed, or combined in some other way to be considered a single product.
Is UDI (Unique Device Identification) applicable for medicinal products incorporating medical devices?
UDI-related MDR duties are not necessary for a DDC (Drug Device Combination) covered by the regulation of the medicinal products. Therefore, the package of such a DDC should not be subjected to a device part-related UDI. Additional information is provided in the MDCG 2019-2 guidance on the application of UDI rules to device-part of products referred to in Articles 1(8), 1(9) and 1(10) of Regulation 2017/745. When a DDC’s device component is CE-marked, the integral DDC’s product labelling should adhere to the labelling specifications for medicinal products as specified in the QRD (working group on Quality Review of Documents) templates. There is no need to remove a UDI that has already been physically marked on a device part. The labelling or outside packaging of the pharmaceutical product should not include the UDI.
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.
A notified body is an independent organisation designated by an EU country to assess the conformity of products before being placed on the market. Under EU MDR 2017/745, the notified body is also called a conformity assessment body.
A conformity assessment body (CAB) or a notified body is responsible for carrying out the conformity assessment procedure mentioned in the applicable Regulation.
What does a notified body do?
A notified body assesses whether the product conforms to the requirements set out in the legislation using a conformity assessment procedure. Some essential activities under the procedure include testing, certification, and inspection.
A notified body is a third-party organisation that works for the benefit of manufacturers. The list of Notified bodies is updated on the NANDO website (New Approach Notified and Designated Organisations).
A notified body also has a list of requirements that need to be fulfilled to carry out conformity procedures.
What is a conformity assessment?
Any legal manufacturer of a product can place their products if the product meets all the requirements. Before a product may be sold, it must undergo a conformity evaluation.
The primary goal of the European Commission is to help ensure that dangerous or otherwise non-compliant items do not enter the EU market.
A conformity assessment is done
Before a product is released on the market, its conformance is evaluated.
To demonstrate compliance with all regulatory standards, which include testing, inspection, and certification.
For each product, the applicable product regulation specifies the method of conformance.
In turn, a conformity assessment guarantees the following:
The product about to be placed on the market meets all legal standards.
The procedure ensures the confidence of consumers, public authorities, and manufacturers regarding the conformity of products.
How is the conformity procedure done?
Product legislation describes the conformity assessment method for each individual product. In the case of medical devices, EU MDR 2017/745 is the latest applicable legislation.
Manufacturers may choose between different conformity assessment procedures, if applicable. The manufacturer may carry out the assessment. If the appropriate legislation requires it, a conformity assessment body is involved in the conformity assessment process.
At the end of the conformity assessment, a CE certificate is issued, which implies that the medical devices conform to the requirements set out in the applicable Regulation.
This ensures that the manufacturer can freely market the device in the EU. The CE certificate and the device bear the CE marking adjacent to which the number of the notified body that issued the same is present.
CE marking with the number of the Notified body
Requirements to be met by Notified Bodies
Annex VII of EU MDR 2017/745 mentions the requirements for Notified bodies. Some general requirements like organisational structure, impartiality and confidentiality are mentioned. More specific requirements include the following:
Maintenance of an appropriate QMS
The Quality Management System must be established such that management structure, documentation, policies and objectives are clear. The QMS must ensure that good documentation practices are followed. This includes control of documents and records, continuous reviews and frequent audits.
Resource requirements
Notified bodies must be capable of carrying out all tasks assigned to them under the Regulation with the utmost professional integrity and competence in the specific field, whether done by notified bodies themselves or on their behalf and under their supervision. The personnel responsible for the tasks carried out by notified body must have suitable qualifications and adequate expertise.
Process requirements
The notified body must have documented processes and sufficiently detailed procedures in place for the conduct of each conformity assessment procedure for which it is designated, including the individual steps from pre-application activities to decision-making and surveillance and considering, as necessary, the respective device specification.
FAQs
Do all EU countries have a notified body?
No. Some countries do not have a notified body. The complete list of notified bodies under EU MDR is provided here. Some examples of notified bodies include TUV, SGS and BSI. These notified bodies do not have a presence in each European country, but the certification they provide is accepted elsewhere.
How do I choose the best-notified body for my devices?
While there is no one best-notified body, a couple of things should be considered when choosing one. Cost of the conformity assessment procedure, response, and geographic location of the notified body. Furthermore, choosing a notified body that also allows the MDSAP certification is considerable if the device is marketed to countries like the US, Canada, and so on.
PMCF is a continuous process that updates the manufacturers’ clinical evaluation during the Post-market surveillance. They must collect user feedback, clinical data, and all other clinical experiences.
This article provides an overview of the design, implementation, and appropriate use of PMCF studies. these are an integral part of PMS reports.
There may be some data limitations during the premarket phase, like the duration of the investigation and the number of subjects involved or restrictions in the applicability of clinical data. Also, a complete diagnosis of all the potential risks and benefits is impossible during the premarket phase.
Post Market Clinical Follow-up (PMCF) is a study to answer specific questions like safety, clinical performance, or effectiveness of the medical devices according to their labelling from the routine use of the clinical practice.
PMCF studies address the uncertainties in the benefits or risks of medical devices like:
Unanswered questions about long-term safety, performance, and effectiveness of the clinical data
Novel/Innovative technologies used in the design, materials/products, and principles of the medical devices
Uncertainties in generating results from the study population to other populations like adults to children
Urgent market access due to emergencies like pandemics
Rare adverse events which can be identified using large datasets
Adequacy of mitigation
PMCF studies/reports must be conducted based on applicable laws, regulations, and ethical requirements. The personal information about the patient should be very confidential, and the documents must be approved with ethics committee under proper consent. Specific guidance and standards to be followed like:
Clearly stated objectives
Scientifically sound study design
Appropriate study plan
Design and Implementation
Factors that need to be considered during the design of PMCF studies:
The study settings such as locations must be clearly described
Study population like inclusion and exclusion criteria should be clearly described
Comparison/Control groups should be justified
The sample size should be clearly stated
Adverse events, risk factors, confounding factors and all other measures should be identified
To minimize the loss, frequent follow-up of the type and duration of the patient
Statistical analysis like missing data and modifications should be clearly described
Factors to be considered during the implementation of the PMCF study plan:
Data collection: Data should be evaluated according to the validated measurement methods
Quality control: Ensure good quality, proper training, selection, and supervision should be performed
Final report and interpretation: Demonstrating conclusion based on the original objectives or hypothesis
Based on all the above factors, there might be changes that impact the clinical evaluation and risk management process. Hence, the medical device must be reassessed to comply with the essential principles / general safety performance. Such assessments may include:
PMCF studies state that the data source collected from real-world clinical experience is essential. Examples of such data are as follows:
Patient-generated health data: Patient report outcome or health data collected from the patient, family members or their caregivers
Device registry: A system that collects data, results and the population that got exposed to that medical device
Health record: The medical record that the health care provider has maintained over the period
Administrative data: Data like health insurance claims
Survey data: Data collected by the survey from health professionals, patients/customers
Bias and Confounding
Potential bias is a deviation caused by overestimating or underestimating the treatment effects. Confounding is the distortion that occurs when the study group differs from the other factors. Some methods to control bias in PMCF study include:
Appropriate selection of study population
Use of validated survey instruments
Training the staff according to the standard
Avoid loss during the follow-ups
Selection of proper statistical data
FAQs
How is the PMCF study different from the clinical investigation?
PMCF, as it states, is the post-market study conducted for a CE-marked device to evaluate the device’s safety performance, whereas the clinical investigation is performed for a device to be CE-marked.
How much importance is given to a PMCF in the MDR?
PMCF is understood to be a continuous process in the device life cycle, and every class of device must have a PMCF plan established to proactively collect and evaluate the clinical data. The analysis of a study must be documented in the PMCF evaluation report, which is a part of the CER and the technical documentation.
What could go into a PMCF?
· Complete clinical data with user feedback · Any specific methods include during the PMCF study or survey · Clinical Evaluation Report (CER) · Address any harmonized standards used in the PMCF plan · PMCF objectives
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.
The MDR reinforces the clinical data and evaluation process (article 61 and Annex XIV), and the manufacturer must confirm the device’s conformity to fundamental health and safety requirements using reliable clinical data and evaluation.
The clinical evaluation establishes the device’s safety and capacity to fulfil its intended function. It also evaluates adverse side effects and determines whether the benefit-risk ratio is acceptable.
Manufacturers must plan, carry out, and document a clinical evaluation in line with Article 61 and Part A of Annex XIV.
Clinical data for the medical device are created, compiled, examined, and ultimately evaluated through a systematic and organised process called a clinical evaluation.
The Clinical Evaluation Report (CER), which the manufacturer uses to show that the medical device complies with the general safety and performance requirements specified in Annex I of the MDR, is the end result of the clinical evaluation.
The Clinical Evaluation Report (CER) is an essential component of a manufacturer’s quality management system and an essential component of the technical documentation for the medical device (MDR Article 10 (3)). It must be actively updated on a regular basis utilising information from the post-market clinical follow-up and post-market surveillance of the medical device (PMCF).
Thus, clinical evaluation is a continuous procedure throughout a medical device’s life cycle.
The Objective of Clinical Evaluation
The clinical evaluation aims to show that the medical device can be used as intended while still being safe and effective, including in terms of its clinical advantages.
The clinical evaluation can also be used to reevaluate risks and find previously overlooked hazards or dangers. The acceptability of hazards must be reevaluated by manufacturers using the most recent clinical evidence.
The objectives of the clinical examination include:
The product’s use for its intended purpose under normal circumstances demonstrates conformity with the general safety and performance requirements listed in Annex I of the MDR
Evaluating or excluding undesirable side effects
Proof of the validity of the risk-benefit ratio
Proving the makers’ medical claims.
Alternative product methods and technologies that can be used in place of the treatment being evaluated are evaluated and documented as part of the clinical study.
The clinical evaluation must ensure that the tested product is not worse than the potential substitutes. The clinical evaluation needs to describe and assess the state of the art.
When assessing state of the art, clinical benefits, safety, and performance should be taken into account. When designing and producing their products, medical device makers must take the latest technological advancements into account.
Clinical Evaluation Data
Clinical information gathered while using the medical device forms the basis of the clinical evaluation. The following are some potential sources for these:
Clinical trial(s) conducted by the manufacturer of the medical device
Clinical trial(s) or other research on a known similar product from the scientific literature
Data from post-market surveillance (PMS) are clinically significant, particularly from post-market clinical follow-up (PMCF).
Reports regarding additional clinical trials using the product under review or a comparable product that has been published in the peer-reviewed scientific literature
Manufacturers must consider preclinical data in addition to clinical data when making their clinical evaluations.
For instance, this comprises the outcomes of the following tests: Testing for biocompatibility, electrical and mechanical safety electromagnetic compatibility in accordance with IEC 60601-1-2, usability, software, animal, simulation, and laboratory testing, as well as testing for durability and stability.
For absolutely non-critical products (stand-alone software, dental drills, oral spatulas, etc.) and must be justified by the manufacturer based on risk management, in accordance with MDR Article 61 “Clinical Evaluation” Section 10.
The manufacturer’s claims, the anticipated clinical performance, and the precise interactions the device has with the human body are all taken into account in this explanation.
According to Annex II of the MDR, the manufacturer in this situation must explain in the technical documentation why they believe it is appropriate to show compliance with the general safety and performance requirements based solely on the outcomes of non-clinical test methods, including performance evaluation, technical testing, and pre-clinical evaluation.
Clinical Evaluation Plan (CEP)
A medical device’s clinical evaluation is a continuous process for developing, collecting, analysing, and evaluating clinical data. It is systematic and well-planned.
Manufacturers are required to create and update a clinical evaluation plan in accordance with Article 61 (paragraph 12) and Annex XIV Part A “Clinical Evaluation” of the MDR (CEP).
Basic ideas like the goals and format of the clinical evaluation are already stated in this strategy. The manufacturer establishes the fundamental performance and safety standards that relevant clinical data in the CEP must back up.
With detailed clinical outcome metrics, it outlines the desired clinical advantages for the patient and specifies the intended purpose, intended target groups, and explicit indications and contraindications.
A new required component of the clinical evaluation plan is a clinical development plan (CDP) for organising pertinently planned clinical trials, including a post-market clinical follow-up plan (PMCF plan).
These adjustments give clinical findings more weight. The Clinical Development Plan (CDP) explains how the manufacturer will gather new or extra clinical data through clinical trials or observational studies to solve open “gap analysis” problems at the beginning of the development phase.
Human volunteers are used in clinical trials to assess the clinical effectiveness and safety of medical equipment.
Class III Devices and Implantable Devices
Clinical investigations must always be carried out in the case of implanted devices and class III devices, with the following exceptions:
The already marketed device has been altered by the same manufacturer, who has also shown that the altered device is equivalent to the marketed one.
The notified body has approved of this demonstration, and the clinical assessment of the marketed device is sufficient to show that the altered device complies with the necessary safety and performance requirements.
Additionally, there is no requirement for clinical testing for class III and implantable devices if a manufacturer can show that its product is functionally equivalent to a product that has already been marketed, provided that the notified body has approved the demonstration and the following requirements are met.
The two manufacturers have a contract in place that expressly grants the manufacturer of the second product full access to the technical documentation on a continuing basis, and the original device manufacturer is still in business.
Additionally, No obligation for clinical investigation for Class III and implantable devices:
If the devices have been legitimately marketed under previous directives, the clinical evaluation is supported by enough clinical data, and they adhere to Common specifications where they are available.
Annex XIV
A clinical evaluation must be planned, continually carried out, and documented by manufacturers in order to:
Create and maintain a clinical evaluation plan,
Utilising a systematic, scientific literature study, determine the clinical data that is available that is pertinent to the device and its intended use, as well as any gaps in the clinical evidence;
Evaluate each relevant clinical study’s applicability for proving the device’s performance and safety;
To produce any additional or new clinical data required to address unresolved problems through adequately conducted clinical research in accordance with the clinical development strategy; and
In order to conclude the safety and clinical performance of the device, including its clinical advantages, all pertinent clinical data must be examined.
Equivalence
Equivalence for the EU MDR clinical evaluation must be proven in two distinct ways.
Clinical
Used for the same clinical condition (with equivalent severity and stage of disease).
Utilised for the same medicinal purpose, and utilised for the same intended purposes, and
Utilised at the same body location, and used in a population with similar features (e.g., age, gender, anatomy, physiology, etc.), and not anticipated to produce noticeably differing performances (in the relevant critical performances such as the expected clinical effect, the specific intended purpose, the duration of use, etc.).
Technical
Have similar specifications and properties (e.g., physicochemical properties such as type and intensity of energy, tensile strength, viscosity, surface characteristics, wavelength, surface texture, porosity, particle size, nanotechnology, specific mass, atomic inclusions such as nitrocarburising, oxidability),
Similar design
Used under the same conditions, similar deployment methods (if applicable), and similar operating principles).
Biological
Use the same tools or substances when in contact with the same body fluids or human tissues.
FAQs
What do you mean by clinical evaluation?
A clinical evaluation is a systematic and well-planned procedure used to acquire, gather, analyse, and ultimately evaluate clinical data for a medical device.
What is clinical evidence?
Clinical evidence is defined as clinical data and clinical evaluation results about a device of sufficient amount and quality to permit a qualified assessment of whether the device is secure and provides the expected clinical benefit(s) when used in accordance with the manufacturer’s instructions.
During the conformity assessment, the manufacturer must submit the complete information material (labelling, IFU (Instructions for Use), any promotional materials and other relevant documents), Clinical evaluation plan & report with the available clinical data, which includes (General Safety and Performance Requirements) GSPR, intended target groups and purpose, qualitative and quantitative aspects of clinical safety, performance procedures and other risk factors involved.
The manufacturer must carry out a gap analysis to fill the sufficient data according to the guidelines to meet the safety and performance of the device as in PART A Annex XIV in MDR 2017/745.
A clinical evaluation report (CER) is a technical document submitted by the manufacturer for the conformity of the devices. While updating the clinical evaluation, the manufacturer must consider,
MEDDEV Guidelines & Frequency of Updates
If the device carries any frequency risk during the intended use
If the device needs any changes like clinical sciences, materials, or other evaluation
Consider the data available on the PMCF (Post Market Clinical Follow-up) reports, clinical investigations, and other studies which may influence the frequency of updates.
Changes in the design or procedure
For the showing of equivalency – technological, biological, and clinical criteria must be considered.
Some general considerations during the Updates
PMS (Post Market Surveillance) always gets updated with new data like safety reports, published literature, registries, PMCF reports and other data during the intended purpose.
Those data should be fed into the clinical evaluation periodically. Also, check the benefit-risk ratios, undesirable side effects and other risk mitigations during the updates.
This may result in changes in the risk management reports, IFUs (Instructions for Use) and PMS activities as in PART B Annex XIV in MDR 2017/745.
Before conducting clinical evaluation and/or investigation for all class III and class IIb devices, the manufacturer is permitted to consult the expert panel mentioned in Article 106 MDR to review the manufacturer’s intended clinical development strategy and proposals for clinical investigation.
In particular, the criteria for an appropriate data set for assessment of the conformity of a device, regarding the clinical data necessary for clinical evaluation, physio-chemical characterization, and microbiological, biocompatibility, mechanical, and electrical considerations, shall be made available to the Member States, notified bodies, and manufacturers through the Commission.
The person who performs the clinical evaluation should know
Research Methodology
Information management
Regulatory requirements
Medical writing with a degree from higher education or 5 to 10 years of professional experience
The technology of that device and complete information
Diagnosis and management of medical alternatives with the standards
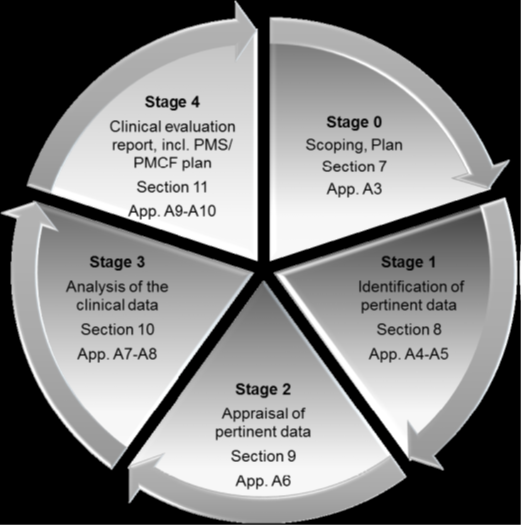
Source: MEDDEV 2.7 Clinical Evaluation (Stages of clinical evaluation)
Stage 0: Define the Scope
Basic and various aspects of the scopes are
Initial device description
Design features, intended purpose, applications of the device and other equivalence
Complete risk management documents which address clinical risks
Any changes in the design, material, or procedures. Also, any changes in IFUs, labels or other promotional materials
PMS aspects like new clinical data or knowledge which may change the state of the art and other aspects
It is crucial to acknowledge that there is a wide range of types, histories, and hazards associated with the technology employed in medical devices.
Since many devices have undergone incremental development or modification, they are not unique. It might be possible to use the clinical reports of an equivalent device’s performance and safety from experience and literature to establish the requirement for clinical data acquired by clinical trials by utilizing the clinical evidence investigation of the experimental device.
Stage 1: Identification of Pertinent data
Manufacturer-generated and stored data
All premarket clinical investigations, risk management activities and PMS programs like PMCF studies, vigilance reports, literature and compliance reports, field safety corrective actions, and other user reports.
Literature Information
Searching the literature reveals prospective sources of clinical data. The following aspects need to be considered.
Identify all the relevant favourable and unfavourable data
Obtain several types of searches like scientific literature databases, internet and non-published data and other literature that has a direct interest
Literature search and other appraisals of clinical data
The literature search must be thoroughly documented so that the methodology can be evaluated critically, and the findings can be confirmed, and the search can be repeated as needed.
Stage 2: Appraisal of Pertinent Data
The appraisal/evaluation plan includes
Criteria for judging the methodological quality and scientific validity of each data set
Standards for establishing the device’s intended use relevance to the clinical evaluation
Standards for weighing each data set’s contribution to the overall clinical assessment
The following section provides examples of factors that can be considered when assessing the methodological quality and scientific validity of the evidence, as in Article 61(3).
Pre- and post-market clinical investigation study plans like adequacy of sample size, random or blinding of patients, adequacy of the follow-up period, serious adverse effects reports and other medical interventions
Some additional aspects like a clinical investigation plan, case report form, audits, regulatory authority approvals, ongoing clinical investigation report which is conducted outside or in the territory and other gap analysis
Data obtained from case studies, patient dossiers, device registries, vigilance data, and other useful data
Data processing is like converting data to a standard format, and statistics
Good clinical practice with all the legal requirements
Sufficient description with proper disclosure of the report
When assessing the relevance of obtained data, we must consider whether the data are meant to directly demonstrate appropriate clinical performance and clinical safety of the device (commonly referred to as pivotal data) or whether they serve an indirect supportive role.
There is no one well-established approach for weighting clinical data due to the variety of medical devices. Instead, the evaluators should choose the best criteria to apply to each evaluation and adhere carefully to these pre-established criteria.
Stage 3: Analysis of Clinical Data
A device’s clinical efficacy and safety should not be demonstrated using data that are not methodologically sound (such as single patient reports).
The evaluators should employ reliable methodologies, conduct a thorough study, decide whether more clinical research or other actions are required, and identify PMCF needs to establish compliance.
During the evaluation, the evaluator should include
Pre-clinical testing
Benefits-risks associated with the patients
IFU correctly addresses risk mitigating methods
Gap analysis like identifying the entire range of products used during evaluation, conditions of use, the number of patients exposed, severity of adverse events, hazard analysis, current standard of care and other profiles
Some additional investigation into missing data and eradicating the issues of compliance.
The clinical evaluation report should have enough details for a third party to read and comprehend it (e.g., regulatory authority or notified body).
The manufacturer must demonstrate compliance with the general safety and performance requirements using clinical evidence, non-clinical data obtained from non-clinical testing methods, and other pertinent documentation.
These materials must also be included in the technical documentation for the relevant device.
Cross-references between the clinical evaluation report’s contents and the pertinent documentation supporting them are required. Which claims are supported by which pieces of data, and which express the evaluators’ opinions should be clear?
With cross-references to the location in the technical documentation from the manufacturer, the report should include references to literature-based data as well as the titles and investigational codes of any clinical investigation reports (if applicable and available).
FAQs
What are CEP and CER?
The clinical evaluation plan (CEP) outlines the devices that will focus on the CER, including their dimensions, intended uses, target population, and patient clinical benefits. Every time brand-new medical equipment is put on the market, the manufacturer must prove that it conforms to all applicable Essential Requirements (ERs) by using the proper conformity assessment methods.
What is the demonstration of equivalence?
When proving equivalence to another device, the MDR stipulates those technical, biological, and clinical features. Although MEDDEV 2.7 Appendix 1 describes these general qualities and aligns them with the MDR Annex XIV Part A (3) requirement, the requirements for each of the three characteristics differ.
What do you mean by similar devices?
Similar devices are devices that fall within the same general device category. The MDR defines this as a group of devices with the same or comparable intended uses or a shared technology that enables them to be categorized in a general way without reflecting characteristics.
What are the available guidelines and guidance documents for clinical evaluation?