Interaction with other medical products and forms of interaction
Pregnancy and lactation If are pregnant, think may be pregnant or are planning to have a baby or are breastfeeding, ask doctor before taking the vaccine.
Effects on the ability to drive and use machines
Undesirable effects
Pharmacological Properties
Pharmacodynamic properties
Pharmacokinetic properties
Preclinical safety data
Pharmaceutical Particulars
List of excipients
Incompatibilities
Shelf life
Special precautions for storage
Nature and contents of container
Special precautions for disposal and other handling
BioSimilaritystudies(In Biosimilar)
Marketing Authorisation Holder
Marketing Authorisation Number(s)
Date Of First Authorisation/Renewal ofthe Authorisation
Date Of Revision ofthe Text
Thailand, governed by the Drug Act B.E.2510. The key points are as follows
1. Legal Basis and Scope
Article 79 of the Drug Act mandates that no drug can be manufactured or imported into Thailand without obtaining marketing authorization from the Thai Food and Drug Administration (TFDA).
Articles 80, 81, and 82 of the Drug Act specify the application dossier requirements, including quality, non-clinical, and clinical information following the ASEAN Common Technical Documents (ACTD) or ICHCTD standards.
2. Application Particulars
The marketing authorization application must include details such as trade name, formulation, pack size, analytical method, label, product leaflet, and other documents as outlined in the Ministerial Regulation.
3. Variation Approval
Any variation to a marketing authorization requires prior approval from TFDA.
4. Procedural Mechanism
The procedures for marketing authorization applications, variation requests, and the issuance of credential certificates align with Ministerial Regulation No.18 (B.E.2525) under the Drug Act B.E. 2510.
5. Role of Drug Committee
Article 10 (1) of the Drug Act assigns the Drug Committee the duty to provide advice or justification for permitting the manufacturing, sale, or import of drugs into Thailand, including their marketing authorization.
6. Focus on Vaccines
The guideline specifically addresses procedural aspects related to the marketing authorization of vaccines.
A vaccine is defined as an immunogen intended to stimulate the immune system for disease prevention, amelioration, or therapy. It may take various forms, including live attenuated preparations, inactivated whole organisms, recombinant DNA-derived immunogens, and more.
Administrative and Scientific
Administrative and scientific advice is available at various stages of the drug development process. It can be sought during initial development, before submitting a marketing authorization application, or in the post-opinion phase.
4 Procedure for Submission of the Application for A TFDA Marketing Authorization Decision
Before submitting an application for marketing authorization to the TFDA, applicants can request a pre-submission meeting. This meeting allows discussions on procedural, regulatory, or legal issues related to the proposed submission.
To request such a meeting, applicants need to use the “Pre-Submission Meeting Request Form.”
1. Before Submission
At least six months prior to submission of an application for Marketing Authorization decision, applicants should notify the TFDA of their intention to submit an application and give a realistic estimate of the month of submission.
In that notification applicants should include:
a draft SPC;
an indication on the number of strengths / pharmaceutical forms / pack sizes (if already known);
if applicable, their intention to present any existing Vaccine Antigen Master File (VAMF) or Plasma Master File (PMF) Certificates
the details of proposed manufacturing sites for the finished product and active substances
a specification of any regulatory issues or difficulties already identified which may require clarification or detailed consideration
2. Submission of the Application
The applicant shall submit the application at the One Stop Service Center (OSSC), TFDA a temporary permit number is issued to the applicant, and verification is completed within 30 calendar days.
3. Dossier to be Submitted
Supporting documents must be submitted in electronic Common Technical Document (eCTD) or other electronic submission platforms as appropriate. If the applicant wishes to use the VAMF or PMF certificate included in the application, the applicant must provide a valid certificate of conformity with the VAMF or PMF in accordance with the EMEA Directive 2003/63/EC Part III and the accompanying assessment report must be provided.
Use relevant VAMF and PMR data. In the case of vaccines containing or consisting of genetically modified organisms (GMOs), the request must be accompanied by documentation from the competent authorities for the intentional release of GMOs into the environment.
In addition, the applicant must provide evidence of the establishment of the applicant and future marketing license holder in the Kingdom of Thailand, as well as documents showing the ability and commitment to perform all the responsibilities required of the marketing license holder, in particular:
• a document identifying the person for pharmacovigilance who will be the contact person for any specific pharmacovigilance issues, together with a curriculum vitae and contact details
• a document identifying the contact person responsible for any quality issues including its contact details
4. Validation by the TFDA
During verification, the TFDA PTL may determine the need for actions related to items such as GMP testing, samples for analysis, GCP testing, communication with environmental authorities, and data integrity.
If TFDA needs additional data, information, or clarification to complete document approval, it will contact the applicant and request to provide such data, information, or clarification within a specified deadline.
4.1. Positive outcome of the validation
If the result is positive, the TFDA has 60 days the applicant must notify in writing that the verification has been completed. Ask external TFDA experts to send 4-7 copies of the document to TFDA PTL.
Applicants must also enter additional data. Information provided during the verification of supporting documents. The evaluation plan adopted by TFDA will be attached to the letter. Confirm positive validation results. Individual arrangements must create a PTL for the copies have.
4.2. Negative outcome of the validation
In the event of a negative validation outcome, where the requested data, information, or clarifications are not provided, the applicant will receive written notification.
If the application cannot undergo validation, the applicant will be given the option to either retrieve the dossier or authorize its destruction by the TFDA. Initiating a new procedure becomes necessary if the applicant plans to submit a complete dossier to the TFDA in the future.
4.3. Management of applications
Data Entry into Tracking System: Once validated, product details are recorded in the TFDA tracking system using a numbering system that provides clear identification throughout the product’s life cycle, including applications for marketing authorization, variations, and transfers.
Identification of Vaccine Applications: Applications for TFDA marketing authorization of vaccines can be identified by the invented name or, if available, the international non-proprietary name (INN)/common name of the active substance(s) combined with the applicant’s name.
For administrative purposes, each application is assigned a core number, consisting of four sections: 1A9000X/year, 2A9000X/year, 1B9000X/year, 2B9000X/year, 1C9000X/year, 2C9000X/year. Here, 1 represents one active substance, 2 represents more than one active substance, A stands for manufacture, B for repack, and C for import.
4.4. Need for samples and sample analysis
No Initial Sample Requirement: Applicants are not required to submit samples of the proposed vaccine at the time of application submission.
TFDA Request for Sample Testing: TFDA encourages applicants to submit vaccine samples promptly for testing at the Division of Biological, Department of Medical Sciences. This early submission helps ensure timely testing.
Test Protocol Specification: The National Control Laboratory (NCL) at the Institute of Biological, Department of Medical Sciences, in collaboration with TFDA, will outline a test protocol.
This protocol includes details such as sample types, sample quantity, batch numbers, specified tests, and the methods and specifications to be used.
Reporting Test Results: Upon completion, the test results are reported to the TFDA Product Team Leader (PTL). These results play a crucial role in the finalization of the TFDA Assessment Report.
How to do medical device registration with the Medicines and Healthcare products Regulatory Agency (MHRA)?
The following steps are followed to register a medical device in the UK market as per the news released by MHRA on 10 Jan 2024.
What can be registered?
Before being sold in the United Kingdom, all medical equipment, including IVDs, specially designed devices and systems, procedure packs, must be registered with the MHRA.
Which regulations must they conform to?
For a device to be placed on the market and registered with the MHRA, it must comply with the Medical Devices Regulations 2002 (SI 2002 No 618, as amended) (UK MDR 2002) as they are implemented in Great Britain.
What are the registration requirements in Northern Ireland?
Devices that you have produced that are Class I, IIa, IIb, or III
Any system or procedure pack that includes at least one medical equipment, whether it be a Class I, IIa, IIb, or III device that you have refurbished or re-labelled under your own name
Custom-made devices
IVDs that you have manufactured and are undergoing performance assessment.
How to register your device to be placed in Great Britain Market?
A single UK Responsible Person must be designated by the manufacturer if their headquarters are located outside of the UK to assume accountabilities for all their medical devices.
Note: Since January 1, 2022, the MHRA registration system has suspended the accounts of any represented manufacturers and any former Authorised Representatives based in Great Britain who have not updated their role to UK Responsible Person. This suspension will continue until the UK Responsible Person has updated their role.
Regulatory Pathway for Clinical investigation creates a set of clinical data that is crucial to understanding the effectiveness of a medical device.
Clinical Investigation is done for previously CE marked devices or for research medical devices. Clinical Investigation is governed by a clinical investigation plan with an objective, methodology, and record keeping.
Top 5 Steps involved in Clinical Investigation
Step 1
Appointment of a Representative- Appoint a legal representative.
Step 2
The application is made electronically via EUDAMED. A single identification number for the clinical investigation is generated. Within ten days of receiving the application, the Member State concerned shall notify the sponsor if the clinical investigation is within the scope of this Regulation and whether the application dossier is complete.
Until the EUDAMED is fully functional, contact the EU Commission authority of the individual member state for the complete set of procedures to be followed. A list of contact persons for Clinical Investigations can be found here.
Step 3
Assessment by the Member States
Conditions based on which the acceptance or refusal of Clinical investigation is mentioned in Article 71 of EU MDR.
Step 4
Proceed with Clinical investigation.
Step 5
Submission of results
Irrespective of the clinical investigation outcome, within one year of the end of the clinical investigation, the sponsor shall submit a clinical investigation report to all the Member States.
Documents required for application
The MDCG guidance document on Clinical Investigation also has the template for the list of documents required and the GSPR. Annex XV Chapter II of EU MDR also contains the complete list of documents required for submission.
The list of documents needed before the application of clinical investigation is as follows:
Name, address, and contact of the sponsor and, if applicable, name, address and contact details of its contact person or legal representative following Article 62(2) established in the Union.
name and other details of the manufacturer
Investigator’s Brochure
Clinical Investigation Plan
Other information
A statement by the natural or legal person responsible for the manufacture of the device under investigation that it conforms to the general safety and performance requirements.
Where applicable, according to national law, copy of the opinion or opinions of the ethics committee or committees concerned.
Proof of insurance cover
Documents to obtain informed consent include the patient information sheet and the informed consent document.
Full details of the available technical documentation, detailed risk analysis or any test reports, shall, be submitted to the competent authority reviewing an application upon request.
Modification of information in Clinical Investigation Plan
Modifications of the clinical investigation plan, investigators brochure, the subject information sheet, and other clinical investigation documentation may or may not be considered substantial modifications. The sponsor or representative must submit a notification of substantial modification following Article 75 of the EU MDR as soon as a clinical investigation is allowed to start.
It is not recommended to submit another substantial modification while the previous assessment is still ongoing. It is also essential to consider if national procedures may apply regarding modifications to clinical investigations.
Reporting Of Adverse Events
The sponsor or representative shall report, without delay, to all Member States in which the clinical investigation is being conducted. Sponsors or representatives should report the following:
any adverse event identified as critical to the results of that clinical investigation.
Any serious or adverse event.
Any device deficiencies that might have led to an adverse event if appropriate action had not been taken.
Any new findings of any event referred to in points 1-3.
Regulatory Pathway for Other Clinical Investigations (If the investigation is conducted not for conformity purpose)
Step: 01
Appoint a Legal Representative who is placed within the European Union. All communications relating to CI will be handled by the legal local representative.
Step: 02
Get an expert panel opinion from the Ethics Committee. This opinion from the Ethics Committee is valid within the entire Member State and the final opinion must not be a negative opinion.
For research purpose investigations, each Member State shall define any additional requirements for such investigations, as appropriate for each Member State concerned.
Hence the Manufacturer must approach each Member state committee contact points to identify their local Investigation requirements.
Step: 05
Conducting Clinical Investigation
Clinical investigations shall be designed and conducted in such a way that the rights, safety, dignity and well-being of the subjects participating in a clinical investigation are protected and prevail over all other interests and the clinical data generated are scientifically valid, reliable and robust.
Clinical investigations shall be subject to scientific and ethical review. The ethical review shall be performed by an ethics committee in accordance with national law. Member States shall ensure that the procedures for review by ethics committees are compatible with the procedures set out in this Regulation for the assessment of the application for authorisation of a clinical investigation. At least one lay person shall participate in the ethical review.
Step: 06
Submit Clinical Investigation Report
Within 1 year of completion of the CI, the Sponsor (manufacturer) must submit the CI report to the concerned Member states.
Source of Information
EU MDR Article 82 states the Requirements for Research only Clinical Investigation (not for conformity purpose)
Article 62 (2) and (3), points (b), (c), (d), (f), (h), and (l) of Article 62(4) and Article 62(6).
FAQs
What are the obligations of a legal representative in Clinical investigations?
The obligation of a Legal representative includes:
Submitting documents to the local authorities.
Notification of clinical investigation.
Reporting any adverse or serious events.
Who is responsible to report adverse events other than during Clinical investigations?
The sponsor is responsible to report adverse events only during Clinical Investigations. In case of general adverse events occurring in the field, the manufacturer is responsible.
What are the supporting documents required for applying Clinical Investigation request?
Mandatory documents:
Cover letter
Application form
Investigator’s brochure
Clinical Investigation plan
Clinical Evaluation plan
Synopsis
Statement of Conformity
Labels (examples)
Description of the arrangements to comply with the applicable rules on the protection and confidentiality of personal data/ personal information
List of General Safety and Performance Requirements
Listed below are as Applicable
Description of the arrangements to comply with the applicable rules on the protection and confidentiality of personal data/ personal information
Risk management documentation
Test reports
Proof of Clinical Investigation Insurance
Suitability of investigational sites and investigation site team
Manufacturer’s Instructions for Use
Suitability of the investigators
Recruitment procedures and advertising materials
Documents to obtain informed consent, informed consent procedure, all written information to participants, payments and compensation of participants
The expert panel in the field of in vitro diagnostic medical devices now accepts submissions from notified bodies for the Performance Evaluation Consultation Procedure | 14 October 2021
To prevent disruption in the supply of these essential healthcare products, the European Commission has proposed a progressive roll-out of the new In Vitro Diagnostic Medical Devices Regulation introducing substantial changes in the regulatory framework for in vitro diagnostic medical devices, such as HIV tests, pregnancy tests or SARS-CoV-2 tests. The length of the proposed transition periods depends on the type of device:
• Class D (High Risk Devices): until May 2025 and 2026 • Class C (High to Moderate Risk Devices): until May 2025 and 2026 • Class B (Moderate to Low-Risk Devices): until May 2027 • Class A (Low Risk Devices): until May 2027
Questions and Answers on the progressive roll-out of the new In Vitro Diagnostic Medical Devices Regulation | 14 October 2021
The European Commission has proposed Questions and Answers on the progressive roll-out of the new In Vitro Diagnostic Medical Devices Regulation (IVDR) to prevent disruption in the supply of these essential healthcare products. The general application date of the Regulation remains 26 May 2022. In particular, the IVD Regulation will apply in full as of 26 May 2022 to CE marked in vitro diagnostic medical devices that do not require the involvement of a notified body (i.e., class A non-sterile devices which represent around 20% of the market) and to ‘new’ in vitro diagnostics (i.e., those not covered by a certificate or a manufacturer’s declaration of conformity issued prior to 26 May 2022).
MDCG: Guidance on classification of medical devices | October 2021
The purpose of this chapter is to provide a general overview on the impact of the classification of medical devices on different aspects of the device compliance with the legal requirements as the classification of medical devices in EU medical device legislation is a risk-based system considering the vulnerability of the human body and the potential risks associated with the devices.
MDCG 2019-6 Rev3: Requirements relating to notified bodies| October 2021
The document presents questions and answers on requirements relating to notified bodies under Medical Devices Regulation (MDR) 2017/745 and In Vitro Diagnostic Medical Devices Regulation (IVDR) 2017/746 under following categories: • Organizational and general requirements • Resources requirements • Process requirements
MDCG: Regulation (EU) 2017/745 – application of MDR requirements to ‘legacy devices’ and to devices placed on the market prior to 26 May 2021 in accordance with Directives 90/385/EEC or 93/42/EEC | October 2021
The MDCG set up an impromptu task-force regarding the application of transitional provisions laid down in Article 120(3) of Regulation (EU) 2017/745 (MDR), the consequential application of MDR requirements to ‘legacy devices’ and to devices placed on the market prior to 26 May 2021 in accordance with Directives 90/385/EEC or 93/42/EEC. The report of the task force was forwarded to all MDCG working groups and MDCG agreed to publish the task-force report as MDCG guidance at its meeting on 19 October 2021.
MDCG: Questions and Answers on repackaging & relabeling activities under Article 16 of Regulation (EU) 2017/745 and Regulation (EU) 2017/746 | October 2021
The document presents questions and answers about obligations introduced by Article 16(2) to (4) under Medical Devices Regulation (MDR) 2017/745 and In Vitro Diagnostic Medical Devices Regulation (IVDR) 2017/746. Article 16(1) of the Regulations outlines cases where the obligations of manufacturers also apply to importers, distributors or other natural or legal persons. Article 16(2) of the Regulations specifies the cases in which certain activities of importers and distributors are not considered modifications of a device, within the meaning of Article 16(1)(c).
Note: Article 16(2), (3) and (4) of the Regulations do not apply to operators subcontracted by the manufacturer (that may also qualify as importers or distributors), who carry out relabelling and/or repackaging activities on behalf and under the control of the manufacturer.
FRANCE
Regulatory developments impacting medical devices that contain cobalt | 01 October 2021
Cobalt, most often alloyed with other metals, is a natural metallic element that is used in the manufacture of many medical devices. As of 1 October 2021, cobalt is classified as a substance: carcinogen 1B, mutagen 2, toxic for reproduction 1B, pursuant to Delegated Regulation 2020/217 of the European Commission but the use of this metal in MDs is not prohibited by this delegated regulation. In accordance with EU MDR 2017/745, devices containing cobalt in a concentration greater than 0.1% by mass fraction (m/m) must now meet the new safety and performance requirements set out in point 10.4 of Chapter II of Annex I. As of 1 October 2021, manufacturers of an MD that contains cobalt must: • justify the use of cobalt; this can be done, for example, by explaining why another material cannot replace it • indicate the presence of cobalt by specific labelling on the MD • provide a leaflet informing of the residual risks
HUNGARY
EU Electronic Application Forms:Electronic application formseAF 1.25.0.0 version | 19 October 2021
The National Institute of Pharmacy and Nutrition (OGYÉI) informed the marketing authorization holders and applicants about the mandatory use of the electronic application forms eAF 1.25.0.0 starting from 1 November 2021 highlighting implementation of the Medical Device Regulation Art 2(1) of Regulation (EU) 2017/745 as the major change in the version.
ITALY
Medical devices: Update of the Q&A document relating to the requirements of notified bodies| 21 October 2021
The document provides clarifications on the requirements and operating procedures for Notified Bodies, authorized by the Authorities responsible for notified bodies to carry out certification procedures for the purpose of CE marking of medical devices, pursuant to Regulation (EU) 2017/745, which allows their placing on the market, putting into service and free movement within the Community in the territory of the European Union. The update of the Questions and answer document mainly provides clarifications on the services offered by notified bodies in the phases that precede the actual certification process in different ways.
Medical devices, classification guidelines | 15 October 2021
The purpose of this document is to provide a general overview on the impact of the classification of medical devices on different aspects of the device compliance with the legal requirements along with the examples of the risk classes to be attributed to different medical devices in accordance with Annex VIII of Regulation (EU) 2017/745.
AUSTRALIA
Discussion paper on potential for mandatory reporting of medical device adverse events by healthcare facilities in Australia | 19 October 2021
The Australian Government supported working closely with healthcare facilities, state and territory health departments to find ways to increase rapid information sharing about medical device safety and effectiveness following the Senate Inquiry into “the Number of women in Australia who have had transvaginal mesh implants and related matters” which highlighted that the number, range, and complexity of medical devices will increase over time.
Submissions received: Proposed refinements to the requirements for medical device patient information materials | 18 October 2021
The TGA thanked respondents who provided a submission to the public consultation related to the implementation of the requirement for sponsors of implantable medical devices and provided patient information leaflets (PIL) and patient implant cards (PIC) about their medical device. The requirements commenced in late 2018, with a gradual implementation over three years. Guidance documents will be updated to address other concerns raised in the consultation, following a decision from the Government on the proposed refinements.
Essential Principles – consent for noncompliance | 14 October 2021
There are Criminal Offences under S41MA and Civil Penalties under S41MAA of the Therapeutic Goods Act 1989, for persons who import, supply or export medical devices that do not meet the Essential Principles for safety and performance. In circumstances preventing compliance to one or more parts of an Essential Principle for a limited period, sponsors may request consent to import, supply or export medical devices which do not meet an Essential Principle. For sponsors that are applying for consent to import, supply, or export medical devices that are non-compliant with EP13A, a specific form has been developed.
Classification of active medical devices (including software-based medical devices) | 11 October 2021
The guidance assists manufacturers of active medical devices, including software-based medical devices, to correctly classify their devices as classification rules are applied according to the manufacturer’s intended purpose and considering how the device works. Manufacturers must consider all the classification rules in classifying their medical device. Where more than one rule applies, the device must be classified at the highest applicable level.
Global Medical Device Nomenclature (GMDN) Terms | 06 October 2021
Global Medical Device Nomenclature (GMDN) Terms are an international naming and grouping convention used to identify and consistently describe medical devices. GMDN Terms are made up of a five (5)-digit numeric Code, a Term Name, and a Definition. Devices are taken to be of the same kind if they have the following characteristics: • the same Sponsor • the same Manufacturer • the same classification • the same GMDN Term • for Class III medical devices, Class AIMD medical devices and Class 4 IVD devices (other than an immunohematology reagent that is a Class 4 IVD medical device), the same unique product identifier (UPI)
The concept of a kind of medical device forms the basis of such things as • conformity assessment • applications for inclusion in the Australian Register of Therapeutic Goods (ARTG) • custom-made medical device notifications • registrations for transition • adverse event notifications
Regulation of software based medical devices | 11 October 2021
The purpose of the guidance is to help manufacturers and sponsors understand how the TGA interprets requirements, and thus indicate how manufacturers and sponsors can comply. Manufacturers and sponsors are encouraged to familiarise themselves with the legislative and regulatory requirements in Australia, and if necessary, to seek professional advice. It is the responsibility of each manufacturer or sponsor to understand and comply with these requirements.
USA
CDRH Proposed Guidance Development | 26 October 2021
The Center for Devices and Radiological Health (CDRH) posts two lists of guidance documents each year that it intends to publish during the fiscal year along with an annual retrospective list of previously issued guidance’s for stakeholder review and input. These lists are: • The A-list: A list of prioritized guidance documents that the Agency intends to publish during the fiscal year. • The B-list: a list of guidance documents that the Agency intends to publish as resources permit during the fiscal year.
CDRH Proposed Guidance’s for Fiscal Year 2022 (FY2022) | 26 October 2021
The Center for Devices and Radiological Health (CDRH) posts two lists of guidance documents each year that it intends to publish during the fiscal year along with an annual retrospective list of previously issued guidance’s for stakeholder review and input. These lists are: • The A-list: A list of prioritized guidance documents that the Agency intends to publish during the fiscal year. • The B-list: a list of guidance documents that the Agency intends to publish as resources permit during the fiscal year.
Notice to Public of Website Location of Center for Devices and Radiological Health Fiscal Year 2022 Proposed Guidance Development | 27 October 2021
The Food and Drug Administration (FDA) is announcing the website location where the Agency will post two lists of guidance documents that the Center for Devices and Radiological Health (CDRH) intends to publish in fiscal year (FY) 2022. In addition, FDA has established a docket where interested persons may comment on the priority of topics for guidance, provide comments and/or propose draft language for those topics, suggest topics for new or different guidance documents, comment on the applicability of guidance documents that have issued previously, and provide any other comments that could benefit the CDRH guidance program and its engagement with stakeholders.
Medical Device Reporting (MDR): How to Report Medical Device Problems| 20 October 2021
Medical Device Reporting (MDR) is one of the Post Market Surveillance (PMS) tools the FDA uses to monitor device performance, detect potential device-related safety issues, and contribute to benefit-risk assessments of these products. Mandatory reporters (manufacturers, device user facilities, and importers) and voluntary reporters (health care professionals, patients, caregivers, and consumers) are required to submit to the FDA certain types of reports for adverse events, product problems, use errors, product quality issues, and therapeutic failures about medical devices. For voluntary reporting, form FDA 3500 may be used by health professionals or consumers.
Regulatory Requirements for Hearing Aid Devices and Personal Sound Amplification Products; Draft Guidance for Industry and Food and Drug Administration Staff; Availability | 20 October 2021
On 7 November 2013, The FDA Reauthorization Act of 2017 (FDARA) directed FDA to update and finalize the draft guidance entitled “Regulatory Requirements for Hearing Aid Devices and Personal Sound Amplification Products”. This updated draft guidance, which supersedes the 7 November 2013 draft guidance, intend to describe hearing aids, personal sound amplification products (PSAPs), their respective intended uses, and the regulatory requirements that apply to these products. This draft guidance is not final nor is it in effect currently.
Medical Devices; Ear, Nose, and Throat Devices; Establishing Over-the-Counter Hearing Aids | 20 October 2021
The Food and Drug Administration (FDA) is proposing to establish a regulatory category for over the counter (OTC) hearing aids and to make related amendments to update the regulatory framework for hearing aids. FDA propose to: • define OTC hearing aids and establish applicable requirements • amend existing rules for consistency with a new OTC category • repeal the conditions for sale applicable to hearing aids • amend the existing labeling requirements for hearing aids; and update regulations relating to decisions on applications for exemption from Federal pre-emption that would become obsolete because of changes to the hearing aid requirements
Surgical Staplers and Staples for Internal Use – Labeling Recommendations Aids | 08 October 2021
The Food and Drug Administration (FDA) is issued the guidance to provide labelling recommendations for surgical staplers and staples for internal use as malfunctions and misuse associated with these devices have resulted in serious adverse events, including deaths. This guidance would help promote the safe and effective use of surgical staplers and staples for internal use by helping manufacturers develop labelling with information about specific risks, limitations, and directions for use of the device.
Agency Information Collection Activities; Submission for Office of Management and Budget Review; Comment Request; Medical Device Labeling Regulations | 08 October 2021
This information collection supports implementation of medical device labelling requirements governed by section 502 of the Federal Food, Drug, and Cosmetic Act (FD&C Act) (21 U.S.C. 352), codified in Agency regulations, and discussed in associated Agency guidance. Medical device labelling regulations in parts 800, 801, 809, and associated regulations in parts 660 and 1040 (21 CFR parts 660, 800, 801, 809, and 1040), prescribe the disclosure of specific information by manufacturers, importers, and distributors of medical devices about themselves and/or the devices, on the label or labelling for the devices, to health professionals and consumers.
Breast Implant Post Market Safety Information | 27 October 2021
FDA’s post market review of breast implant safety and effectiveness includes:
• Providing input and guidance to manufacturers as they work to meet post-approval study enrolment, follow-up, and objectives. • Communicating study status and findings to healthcare providers and patients to help inform them of risks associated with breast implants. • Evaluating breast implant Medical Device Reports (MDRs), especially those related to breast implant-associated anaplastic large cell lymphoma (BIA-ALCL) and Systemic Symptoms in Women with Breast Implants. • Assessing all post market reporting requirements associated with the approved PMA, including manufacturing changes, design changes, and annual reports. When the FDA finds that a manufacturer has significantly violated the FDA’s regulations, the FDA notifies the manufacturer. This notification may be in the form of a Warning Letter.
Labeling for Approved Breast Implants | 27 October 2021
On October 27, 2021, the FDA took several new actions to strengthen breast implant risk communication and help those who are considering breast implants make informed decisions. The FDA issued orders restricting the sale and distribution of breast implants to help ensure that patients considering breast implants are provided with adequate risk information so that they can make fully informed decisions and approved new labelling for all legally marketed breast implants that includes: • Boxed warning. • Patient decision checklist, which must be reviewed with the prospective patient by the health care provider to help ensure the patient understands the risks, benefits and other information about the breast implant device. The patient must be given the opportunity to initial and sign the patient decision checklist and it must be signed by the physician implanting the device. • Updated silicone gel-filled breast implant rupture screening recommendations. • Device description with a list of specific materials in the device. • Patient device card. The FDA expects manufacturers to post the updated device labelling to their websites within the next 30 days and has released updated information on the status of breast implant manufacturer post-approval studies.
FDA Strengthens Safety Requirements and Updates Study Results for Breast Implants | 28 October 2021
The agency issued orders restricting the sale and distribution of breast implants to help ensure that patients considering breast implants are provided with adequate risk information so that they can make fully informed decisions. • the agency issued orders restricting the sale and distribution of breast implants to help ensure that patients considering breast implants are provided with adequate risk information so that they can make fully informed decisions • the agency has approved new labelling for all legally marketed breast implants that includes a boxed warning, a patient decision checklist, updated silicone gel-filled breast implant rupture screening recommendations, a device description with a list of specific materials used in the device and a patient device card • the FDA released updated information on the status of breast implant manufacturer post-approval studies These actions will help patients understand the risks and benefits of breast implants and make more informed decisions about their health.
UK
UK, USA, and Canadian regulators identify 10 guiding principles to be addressed when medical devices use AI or machine learning software| 27 October 2021
The principles cover key elements of GMLP, for example: • having an in-depth understanding of a model’s intended integration into clinical workflow • the desired benefits and associated patient risks • selecting and maintaining training and datasets to be appropriately independent of each other These guidelines may be used to: • adopt good practices that have been proven in other sectors • tailor practices from other sectors so they are applicable to medical technology and the health care sector • create new practices specific for medical technology and the health care sector
Consultation on the future regulation of medical devices in the United Kingdom | 26 October 2021
Medicines and Healthcare products Regulatory Agency (MHRA) is inviting members of the public to provide their views on possible changes to the regulatory framework for medical devices to enable: • Improved patient and public safety • Greater transparency of regulatory decision making and medical device information • Close alignment with international best practice • More flexible, responsive, and proportionate regulation of medical devices This consultation looks at how we might change the law around medical devices in the UK by updating the Medical Devices Regulations 2002 (as amended) (UK medical devices regulations). Two webinars aiming the industry and public were uploaded on the website.
Guidance: Borderlines with medical devices and other products in Great Britain | 01 October 2021
The guidance document covers borderline products with general medical devices as specified in Part II of the Medical Device Regulations 2002 (as amended) and does not cover borderlines with in-vitro diagnostic medical devices or active implantable medical devices.
SWITZERLAND
Patient care in Switzerland at risk| 17 October 2021
After getting the third country status, Switzerland continues exporting medical products under the new EU regulation but the current situation with imports is alarming. By implementing home-made import barriers, Switzerland is endangering the health care of its own population. Swiss MedTech is therefore urgently calling for changes to the national Medical Devices Ordinance.
PAKISTAN
Draft Guidelines for Imports & Exports of Therapeutic Goods (for Views/Comments of stakeholders) | 25 October 2021
The guidance document aim to provide an overview of the requirements, procedures and best practices for imports and exports in compliance with the legal and regulatory requirements for all therapeutic goods including finished pharmaceutical and biological drug products, active pharmaceutical ingredients (APIs) and drug substances (DS), Medical Devices, and Health & OTC Product (e.g. nutraceuticals, herbals, ayurvedic and homeopathic products, biochemic and Chinese products) and their raw materials. The guidelines are meant to: • outline the requirements and documentation for import and export of therapeutic goods • determine the eligibility, who can import or export therapeutic goods • elaborate procedure adopted by DRAP for verification and port clearance • describe the responsibilities of the entities involved in import and export
KAZAKHSTAN
Kazakhstan has introduced the practice of inspection of medical devices before state registration| 18 October 2021
According to Yerken Dautbayev, Director General of NCELS, special attention is paid to the inspection of domestic manufacturers of medical devices, which should occupy a competitive niche and replace expensive imported medical products, provide domestic health care with high-quality medical products within the framework of the Government’s instructions. Since the beginning of this year, experts from the National Centre for Expertise of Medicines and Medical Devices have conducted inspections for domestic manufacturers of medical products and manufacturers of near and far abroad. The results of the inspection are an integral part of the registration dossier of medical products and are considered when forming the results of the examination.
BRAZIL
Deadline for loading instructions for use ends on 31 October | 04 October 2021
ANVISA informed about the deadline closure for the loading of instructions for the use of regularized medical devices on 31 October 2021 before the validity of The Collegiate Board Resolution (RDC) 431/2020. The standard states that companies holding records and notifications of these products must upload their instructions for use in the Medical Device Documentary Repository, available in the Regularized Product Consultations section of the Agency’s website and was in force since 1 November 2020. Companies holding records and notifications published or published after the validity of RDC 431/2020 have up to 30 days to arrange the loading of documents in the repository, after the regularization of the product with the Agency.
LITHUANIA
For the attention of companies manufacturing orthopedic medical devices | 28 October 2021
The State Accreditation Service for Health Activities under the Ministry of Health invites orthopaedic medical device manufacturers to free distance learning for the application of the new Regulation (EU) 2017/745 of the European Parliament and of the Council on the application of medical devices on 18 November 2021, 9.00 on MS Teams by mailing the name of the company or institution intending to participate in distance learning and name and contact details (e-mail, phone number) of the person who will participate in the distance learning on [email protected]
IMDRF DOCUMENTS
Competence and Training Requirements for Auditing Organizations | 20 October 2021
The document applies to recognized Auditing Organizations conducting audits of a medical device manufacturer for regulatory purposes. Adherence to this document and its requirements will help mitigate the risk of inconsistent or ineffective assessments of manufacturers by ensuring that Auditing Organization personnel have the necessary commitment, competence, experience, and training before conducting an audit or undertaking a decision-making function.
Regulatory Authority Assessor Competence and Training Requirements | 20 October 2021
The document applies to Regulatory Authorities conducting assessments of Auditing Organizations. Adherence to this document and its requirements will help mitigate the risk of inconsistent or ineffective assessments of Auditing Organizations by ensuring that Regulatory Authority personnel have the necessary competence and training before conducting an assessment or participating in a decision to recognize an Auditing Organization.
MDSAP Assessment and Decision Process for the Recognition of an Auditing Organization | 20 October 2021
The purpose of the document is to explain the assessment process and outcomes, including the method to “grade and manage” nonconformities resulting from recognizing Regulatory Authorities assessment of an Auditing Organization and, to document the decision process for recognizing an Auditing Organization or cessation of recognition. This document defines: • The process and lifecycle for recognizing, maintaining, or ceasing recognition of an Auditing Organization. • The process of managing, grading, and closure of assessment nonconformities issued to an Auditing Organization • The outcomes of an initial, surveillance, or re-recognition assessment process of an Auditing Organization
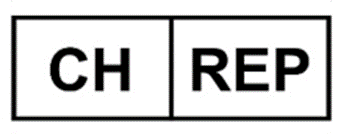
Written, printed, or graphic information appears either on the device itself or on the packaging of each unit or the packaging of multiple devices (Art. 2 point 13 MDR). Timeline for Information on Indication of CH-REP “on the device” or “in the accompanying document.”
The provision for indication of CH-REP comes into effect on 26 May 2021 and is timeline based, applicable to devices of all classes (Class I, IIa, IIb, III).
For imported devices, the CH-REP should be indicated according to the below table:
Device Class
CH-REP Symbol / Indication to be placed on
Class I MDR Device
With effect from 26 May 2021 Until 31-Jul-2023Where: Either on the label (or) On the Document accompanying the device After 31-Jul-2023Where: On the label
Class IIa, IIb, III MDR Devices
With effect from 26 May 2021 Where: On the label
MDD/AIMDD Devices with EU/EEA manufacturer or EC-REP
With effect from 26 May 2021 Where:MDD – On label (or) Instructions for Use (or) In Document accompanying the device AIMDD – On sales packaging (and) Instructions for Use (or) In Document accompanying the device
MDD/AIMDD Devices without EU/EEA manufacturer or EC-REP
With effect from 26 May 2021 Where:MDD – On label (or) Instructions for Use AIMDD – On sales packaging (and) Instructions for Use
“CH-REP” Symbol
Representation / Usage of the “CH-REP” Symbol on the Device packaging
The name of the Swiss AR must be available on the product label.
The Name and Address of the Authorised Representative (AR) must appear adjacent to the symbol.
The mentioned Address must enable provision to contact the Swiss AR with sufficient contact details.
The symbol’s size and name are not defined; however, they must be adequately visible to the naked eye.
Symbols to Use
Where symbols are used, they must confirm to the harmonised standards. Swiss medic rule allows the sole use of the symbol without any description of the symbol explained in the product documentation enclosed.
Other CH-REP Representation(s)
Instead of the symbol, Swiss medic also permits the indication as stated below:
“CH-Authorised Representative”
“CH-REP”
“Authorised Representative for Switzerland”
Rules for Manufacturers from EU/EEA
Manufacturers placed in the EU/EEA also fall under this obligation to appoint their Swiss AR (called CH-REP or Swiss Rep or SAR). It is least expected that a single CH-REP is appointed for devices that belong to the same medical device group or family.
What does “In a document accompanying the device” mean and where should I make this information available?
This information can be affixed to the device or be available separate from the device. Available separate refers to documents such as delivery notes, customs documents, guarantee certificates, invoices, a sticker on the packaging or the instructions for use.
Such documents are made available with the device throughout the supply chain route. It is available until it reaches the distributors and need not be necessarily available to the end customer. Note that this accompanying interpretation differs from the Swiss with EU MDR.
Is the definition of a “document accompanying the device” of Swiss MedDO and Europe’s MDR 2017/745 the same?
No, they are not the same. The interpretation of document accompanying the device definition differs from Swiss MedDO and EU MDR.
In what languages must the medical device product information such as Labelling and Instructions for Use be written for the Swiss market?
It must be written and made available in all three official languages of Switzerland (German, French and Italian). Symbols established by using technical standards may be used to replace the written statements.
What are the other obligations of a Swiss AR / CH-REP?
A detailed obligation/responsibilities of a CH-REP are available in our article Obligations of the Swiss Authorised Representative.
Who are the Economic Operators required to be registered with Swiss medic?
Manufacturers (or) their Swiss Authorised Representative must register themselves within three months of placing the device for the first time in the Swiss market.
During registration, they must cater information as per Part A of Annex VI EU MDR 2017/745. Further obligations and registration modalities are governed by Article 30 and Article 31 of EU MDR 2017/745