

Quality Management Systems (QMS) are defined by the MDR as formalised systems that document processes, responsibilities, and procedures to ensure and continually improve the standard of company activities.
To obtain CE marking and market devices in European Union, an effective Quality Management System that complies with EU MDR 2017/745 is must.
Article 10 of EU MDR 2017/745 explains the general obligation of manufacturers to place their products in the EU market. Article 10 of the EU MDR 2017/745 also comprises the requirements for implementing and maintaining a Quality Management System capable of addressing particular issues to ensure device quality and safety.
Manufacturers of devices are expected to establish, document, implement, maintain, and keep up to date all the device documents to continually aid the improvement of the quality management system.
Ready to Streamline Your Regulatory Compliance?
Join hundreds of companies who trust OMC Medical for their regulatory needs. Get expert guidance and ensure compliance across all markets.
Call Now +44 208 066 7260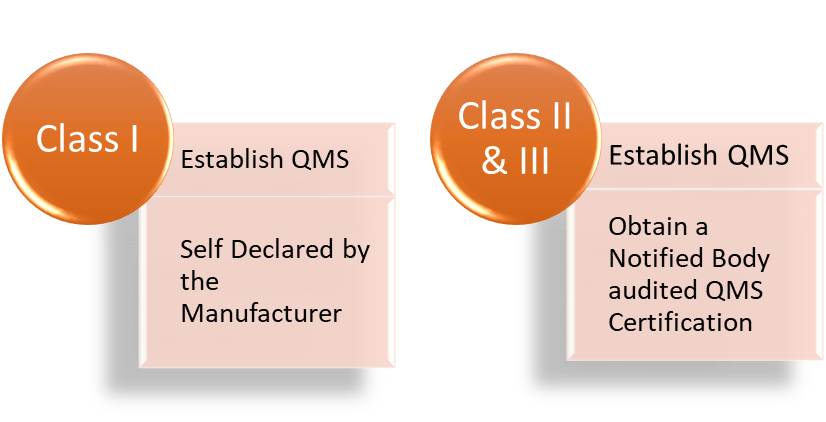
Quality Management System (QMS) must address at least the following:
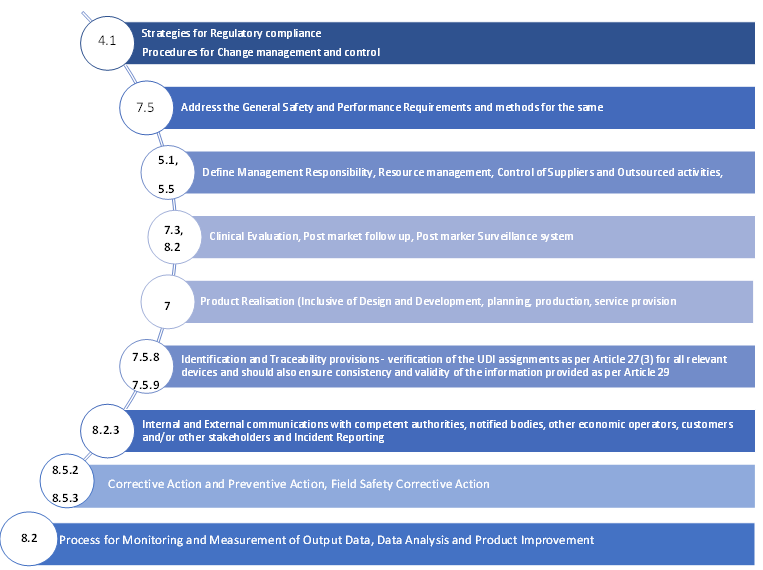
- A strategy for regulatory compliance, which includes conformity assessment procedures and procedures for change management and control for the device
- The system must consist of general safety and performance requirements and methods to address the same
- The system must define the management’s responsibility
- The system must include resource management methodologies which should contain information on the selection and control of suppliers and sub-contractors
- The system must include risk management methodologies as per Section 3 of Annex I
- The system must include clinical evaluation data along with post-market clinical follow-up (PMCF) as per Article 61 and Annex XIV
- The system must include data on product realization, which should involve information such as planning, design, development, production, and service provision
- The system must ensure the verification of the UDI assignments as per Article 27(3) for all relevant devices and should also ensure consistency and validity of the information provided as per Article 29
- The system must contain data on the setup, implementation, and maintenance of a post-market surveillance system as per Article 83
- The system must include communication protocols with competent authorities, notified bodies, other economic operators, customers and/or other stakeholders
- The system must include methodologies for reporting serious events and field safety corrective actions in the context of vigilance
- The system must also include methodologies of management of corrective actions and verification of their effectiveness
- The system must include data on processes for monitoring and measurement of output data, data analysis and product improvement
Mapping of ISO 13485:2016 clauses to the MDR QMS requirements
FAQs:
How will a QMS be assessed under the EU MDR?
The quality management system of a manufacturer will be evaluated as part of the EU MDR Annex IX to XI- conformity assessment procedures. A Notified Body will do conformity evaluation for all devices except Class I.
Can ISO 13485:2016 QMS certificate holders bypass or claim EU MDR compliant QMS by themselves?
No, to be compliant with EU MDR the manufacturer is required to obtain a QMS assessment w.r.t Article 10(9) by a notified body in case of Class II & III. For a class I, the manufacturer must establish a corresponding QMS and include the same declaration in the self-declared DoC
Is 13485:2016 mandatory for MDR in addition to Article 10(9)?
No, the ISO 13485:2016 is not mandatory. However, the Quality Management System ISO 13485:2016 ensures entry to various global markets.
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.





