Obligations of the Switzerland AR Responsibilities
1. Verify the DoC and technical documentation have been drawn up and where necessary conformity assessment procedure has been carried out.
2. Keep available a copy of the technical documentation, the declaration of conformity and, if applicable, a copy of the relevant certificate, including any amendments and supplements, at the disposal of competent authorities for a period as defined in the section «Records» of this agreement.
3. Register itself on the electronic system «EUDAMED» and obtain the Single Registration Number (SRN)
4. Verify that the manufacturer has complied with the registration obligations in EUDAMED by entering the UDI core data elements (as per Article 27 of (EU) 2017/745) and has assigned and entered in Eudamed the Basic UDI-DI Information of the devices (as per Article 29 of (EU) 2017/745).
5. In response to a request from the competent authority, provide the competent authority with all the information and documentation necessary to demonstrate the conformity of a device, in an official language determined by the competent authority.
6. Forward to the manufacturer any request by a competent authority for samples, or access to a device and verify that the competent authority receives the samples or is given access to the device.
7. Cooperate with competent authority on any preventive or corrective action taken to eliminate or, if that is not possible, mitigate the risks posed by devices.
8. The AR is equally liable for any defective device in the Swiss market when either of the parties does not fulfil their obligations in accordance with the Ordinance. Therefore, it is important to consider the inclusion of AR in the corresponding processes and management reviews as deemed necessary.
9. Immediately inform the manufacturer about complaints and reports from healthcare professionals, patients and users about suspected incidents related to a device.
10. AR shall ensure that it has permanently and continuously at its disposal at least one the person responsible for regulatory compliance who possesses the required expertise (as per article 52 of MedDO / article 15 of (EU) 2017/745) regarding the regulatory requirements for medical devices in Switzerland. The PRRC shall comply with its responsibilities as defined in the Ordinance.
11. The PRRC of AR is responsible to verify the quality functions of the manufacturer (if conformity of the device is checked as per the QMS, the PMS obligations, reporting obligation of incidents, issuance of the signed statement confirming the safety of the investigational device). This could be facilitated by allowing the PRRC of AR to have access to the QMS documentation. For example, the PRRC of AR could be part of the internal audit of the manufacturer.
12. The AR is legally liable for defective devices on the Swiss market on the same basis as, and jointly and severally with, the manufacturer when the manufacturer has not complied with its obligations according to article 10 of (EU) 2017/745
1. Who is responsible for medical device regulation in Switzerland?
The Swiss Agency regulates medical devices for Therapeutic Products (Swiss medic)
2. Which current Switzerland medical device regulations need to be followed by stakeholders?
From May 26th, 2021, Stakeholders need to follow the revised Medical Devices Ordinance. (MedDO) and a new Ordinance on Clinical Trials with Medical Devices (CTO-Medd).
3. What is the EU- Switzerland mutual recognition agreement (MRA)?
EU- Switzerland mutual recognition agreement (MRA) is a crucial agreement between the EU and Switzerland. It allows the bilateral trade of several goods like medical devices, heavy machines, vehicles, etc. This MRA allowed mutual recognition of conformity assessment certificates between Europe and Switzerland.
4. What is the status of the EU-Switzerland mutual recognition agreement (MRA) for medical devices?
MRA is not updated on May 26th, 2021. As a result, the new medical device regulation of the European Union, i.e., EU MDR 2017/745, is not included in MRA. MRA has previous directives 90/385/EEC and 93/42/EEC in the medical chapter.
5. What are the consequences of this MRA status on the Swiss manufacturers/authorised representative who wants to place their products on the EU market?
From now on, Switzerland will be treated like a third-world country by the European Union. Manufacturers & authorised representatives based in Switzerland will need to appoint an EU Authorised representative in the EU and followed EU-MDR 207/745 to place medical devices on the EU market. Medical devices will need to undergo conformity assessment procedures by Notified Bodies in the EU to get CE certificates. All the existing CE certificates based on directives 90/385/EEC and 93/42/EEC under the MRA will not be considered valid in the European Union.
6. Which regulations do I need to follow in order to place my device on the swiss market?
Manufacturers will need to follow the revised Medical Devices Ordinance (MedDO) to place their products in Switzerland.
7. What are MedDO requirements?
Primary requirements of MedDO include the appointment of Switzerland authorised representative (Switzerland AR) for manufacturers based outside of Switzerland, economic operators registration and continuous reporting of severe incidents to Swissmedic, UDI assignment, and device notification.
8. Who needs to register with Swissmedic?
Economic operators (manufacturers, importers, authorised representatives placed in Switzerland) need to register themselves with Swissmedic in the first three months of
placing the device in the market. After successful registration, they will be assigned a Swiss Single Registration Number (CHRN). The Swiss Single Registration Number (CHRN) is a unique identification number to identify. Economic operators who already sell products before May 26th, 2021, in accordance with MDR 2017/745 & IVDR 2017/ 746, registrations in accordance must be completed by November 26th, 2021.
9. What is the process of economic operator registration with Swissmedic? What are the registration fees?
The economics operator needs to submit the information to Swissmedic with the help of a registration form: Application registration CHRN. Swissmedic will review the information. Once it is successfully reviewed, the economic operator will receive CHRN. For the assignment of CHRN, fees will be based on the amount of review work done by Swissmedic per application. It is CHF 200 per hour.
10. What Information is required by Swissmedic for economic operators registration?
Name of the registrant, contact details, details of the person responsible for regulatory compliance, commercial Register extract, For Swiss-domiciled, authorised representatives to mandate from a manufacturer outside Switzerland is required by Swissmedic.
11. Who can be appointed as Switzerland AR? What is the deadline for appointing Switzerland AR?
A Swiss authorized representative (Swiss AR) is a legal person that needs to be appointed by a manufacturer established outside of Switzerland to place the products on the Swiss market. A written mandate/letter designating an authorised representative for a medical device manufacturer is mandatory. Any legal entity or natural person situated in Switzerland can act as Switzerland AR. Switzerland AR is responsible for product safety and will be held liable for product defects. Manufacturers can appoint only a single Authorised representative in Switzerland.
Timeline to appoint Switzerland AR:
12. Which devices need to be notified to Swissmedic? What is the process?
The following device needs to be notified to Swissmedic before placing it on market:
13. What is the timeline to assign UDI? For placing UDI- carriers on the labels of the device deadline for implantable devices and
class III devices are 26th May 2021, Class IIa and class IIb devices are 26th May 2023 and for
class I devises it is 26th May 2025.
14. Will the Swissmedic accept CE certificates? CE certificates from notified bodies placed in EU/EEA countries will be valid in Switzerland until and unless the applied conformity assessment procedures comply with the Swiss requirements meet, and they are issued by a notified body that has an equivalent qualification as described in MedDO.
15. What are the regulations for the In vitro diagnostic devices? In vitro, diagnostic medical devices regulations are in the final step. They will be published as a separate ordinance on in-vitro diagnostics (IvDO).
As the Institutional Agreement is missing (InstA) is missing, the EU has not updated the Mutual Recognition Agreement (MRA)
No barrier-free access to the EU internal market
Swiss has been downgraded to the third country
Swiss manufacturers have stricter requirements when exporting their medical devices to the EU.
The Swiss MedTech industry will have:
The administrative cost of CHF 114 million initially
CHF 75 million annually
Cost represents 2% and 1.4% of total export volume (CHF 5.2 billion) to the EU
Switzerland Market Situation – Downside
Non-EU manufacturers will not want to have an HQ based in Switzerland
Swiss Start ups will move to the EU going forward for ease of trade
Swiss has lost the advantage of an Economically important branch of industry with 63,000 employees and 1400 companies.
Switzerland- Medical Device Regulations
Medical devices are regulated by: the Swiss Agency for Therapeutic Product
Manufacturers Outside Swiss
After 26 May 2021, foreign manufacturers must appoint a Swiss authorised representative in order to sell their devices in the market.
An agreement between the manufacturer and the AR is needed.
Who is a Swiss AR
– Proxy to the foreign manufacturer
– Responsible for product safety
– Liable for product defects
– contact person for Swiss authorities
– can be a legal entity or a natural person
– must have access to PRRC
Roles of Swiss AR’s PRRC
Same as the role of PRRC for pharmaceutical products
It is mandatory for an AR to appoint PPRC or delegate this to an external service provider
Assumes a central role in PMS surveillance and in the control of medical devices
He/she is personally responsible for the compliance with the duties incumbent on the AR and can be held criminally liable for the violation
Liability of a Swiss AR
According to the swiss Product Liability Act, the manufacturer is responsible for any product defects.
Swiss Ordinance on medical devices extends liability to Swiss AR
AR is jointly and legally liable for any product defects.
Liability of an AR cannot be waived by contract.
AR and the manufacturer are obliged to take liability insurance with sufficient coverage.
The manufacturer, however, may undertake to pay the insurance costs for the authorized representative.
AR should do due diligence before signing the agreement with the manufacturer.
Manufacturer Responsibilities
The centre of the responsibilities is defined by law.
Prove and document conformity of his product, including correct classification.
The introduction and maintenance of a QMS system is his responsibilities.
AR is dependent on the manufacturer to fulfil its obligations.
The manufacturer must be contractually obliged to guarantee the authorized representative full compliance with his legal duties.
Legal duties include- CE mark, UDI, IFU, and Translation into Swiss languages.
Manufacturers must supply the AR with extensive documentation.
The manufacturer is obliged to retain the technical documentation, declaration, and certificate of conformity, including amendments and supplements for 10-15 yrs.
AR to keep commercial records for 10 yrs.
Notifications
Class I
Custom made devices
Systems and procedure packs
Devitalised human tissue
Repacked or relabeled medical devices
Medical devices manufactured and used in health institutions in the Swiss
IVD and self-testing IVDs, other IVDs
IVD manufactured in house
UDI- CHRN
The Swiss Single Registration Number (CHRN): Unique identification number
Swissmedic assigns CHRN to Swiss manufacturers, authorized representatives and importers
Until the MRA (Mutual Recognition Agreement) is updated, Swissmedic is unable to assign a European Single Registration Number (SRN) via EUDAMED for economic operators who are domiciled in Switzerland.
Registration with Swissmedic is necessary for CHRN
Economic operators must register within three months of placing their first product on the Swiss market.
Update for Manufacturers in EU & Swiss
Registration and notification obligations do not run via EUDAMED but via Swissmedic.
Economic operators who have already placed products on the market before 26 May 2021 in accordance with the MDR and IVDR must complete their registration by 26 November 2021.
Access to the technical documentation may be provided either by keeping a copy available at the authorised representative or by contractually guaranteeing that it will be handed over to Swissmedic upon request within 7 days.
The validated Summary of Safety and Clinical Performance (SSCP) is not uploaded by the Notified Body in EUDAMED but published by the manufacturer, for example on its website.
For manufacturers outside Switzerland:
Need to appoint Swiss AR, update the labelling for MDR products & follow third-country requirements
Timeline to appoint swiss AR:
Swiss Manufacturer exporting to EU:
For MDR regulated devices & class I devices: Follow third country requirements from 26th May 2021
For MDD certified devices: No third country requirements, they will be regulated under MRA, free access to EU market as before until May 2024.
MDCG Position Paper on the Implementation of UDI requirements for contact lenses, spectacle frames, spectacle lenses & ready readers| 27 May 2021
The position paper on the implementation of UDI requirements for contact lenses, spectacle frames, spectacle lenses by MDCG provides clarification on the implementation of UDI requirements from 26 May 2021. For contact lenses, a specific UDI assignment solution is under development, and it might be extended to spectacles frames. For spectacle lenses and ready readers, a specific UDI assignment solution is agreed upon. Its practical application is yet to be finalized. Position paper also details the UDI and device registration timeline in EUDAMED.
Information notice on the status of the EU-Switzerland Mutual Recognition Agreement for Medical Devices | 26 May 2021
European commission has issued a notice to stakeholders about status of EU- Switzerland Mutual Recognition Agreement (MRA) for medical devices. MRA is one of the key agreements between the EU and Switzerland, facilitating bilateral trade for machinery, motor vehicles and medical devices. Up until 26 May 2021, medical device chapter of MRA facilitated trade of medical devices between EU and Switzerland. As the MRA is not updated following are the after-effects that stakeholders should be aware of:
• Switzerland will be treated as third world country. • No mutual recognition of conformity assessment results in EU and Switzerland. • Need of authorized representative in both EU and Switzerland.
Questions & Answers: Application of Regulation on Medical Devices – EU rules to ensure safety of medical devices | 26 May 2021
European commission published the set of Q&A for application of regulation on medical devices. These Q&A explains aspects of EU-MDR like the need of new medical device rules, its benefit to the patient, safety aspects of rules, role of notified bodies, high risk devices scrutiny, rules on reprocessing single use devices & products without an intended medical purpose, EUDAMED
EU-MDR comes into force | 26 May 2021
From 26 May 2021, new EU medical device regulation (EU- MDR) comes into force, establishing robust regulatory framework for medical devices ranging hip replacements to sticking plasters. EU – MDR
improves the quality, safety and reliability of medical devices, strengthens transparency and information for patients, enhances vigilance and market surveillance.
Clinical investigation application/notification documents | 21 May 2021
MDCG published a guidance document on application to be submitted by sponsor of clinical investigation. This guidance lists down the documents which needs to be included in the application/notification to the Member state in which clinical investigation will be conducted. It includes documents such as application/notification form, supporting documents, General safety and performance requirements, standards etc. This application should be provided with documentation referred to Annex XV of EU- MDR 2017/745. The templates of the application and necessary document is also provided in the guidance.
Launch of EU UDI Helpdesk| 18 May 2021
EU UDI Helpdesk is live from 18th May 2021. The UDI system is new requirement introduced by EU-MDR for identification of medical device. The UDI Helpdesk will give support to economic operators in the UDI system implementation requirements. These requirements are assignment of UDI, labelling and registration of devices. The support will also include for the use of European Medical Devices Nomenclature (EMDN). UDI helpdesk has definitions and basic information of MDR, specific questions related to MDR, EUDAMED- related questions, various dates & deadlines of requirement, links, and documents specified by EU MDR.
Notice to manufacturers and authorized representatives on the impact of genetic variants on SARS-COV-2 in vitro diagnostic medical devices | 17 May 2021
MDCG releases a notice for manufacturers & authorized representatives of in vitro diagnostic medical devices (IVDs) with the intended purpose to detect and/or quantify markers of SARS-CoV-2 infection. Notice emphasis on the duties of manufacturers to ensure the safety of the device. manufacturers should continually assess the impact of different new genetic variants on the IVD, also make sure that risk associated with the use of IVD must be accepted when it is weighed against benefits to the patients. Manufacturers also need to make sure that device conforms to essential requirement mentioned in Directive 98/79/EC on IVD. All the incident regarding devices should be reported to competent authority by manufacturer.
Guidance on harmonized administrative practices and alternative technical solutions until EUDAMED is fully functional| 04 May 2021
The revised guidance on the harmonized administrative practices and solutions until EUDAMED is fully functional includes guidelines for economic operators, competent authorities, other commissions to fulfil EU-MDR requirements related to EUDAMED. It proposes alternative solutions to submit to competent authority by responsible actors. It includes provisions such as but not limited to registration of devices, UDI system, summary of safety and clinical performance, European database on medical devices, Clinical evaluation consultation procedure for certain class III and class IIb devices. y. This guidance also foresees the launch of EUDAMED for May 2022, the date of application of Regulation (EU) 2017/746 on in vitro diagnostic medical devices (IVDR)
UK MHRA
Updated list of medical devices given exceptional use authorisations during the COVID-19 pandemic |27 May 2021
During pandemic to ensure the supply of medical devices in the UK, MHRA publishes list of manufacturers and devices that are granted exceptional use application. This list gets updated by MHRA every week. Medical devices granted an exceptional use authorisation can be sold to the NHS and within the social care.
Guidance: Notify the MHRA about a clinical investigation for a medical device| 25 May 2021
The UK Government has updated the guidance of Notifying the MHRA about a clinical investigation for a medical device. From 26th May 2021, Northern Ireland will be following EU MDR. The update includes technical details for clinical investigations in Northern Ireland. Application should be submitted to MHRA in line with the requirements of EU MDR 2017/ 745. It also provide guidance for the post market studies involving sites in Northern Ireland.
Guidance: Register medical devices to place on the market|24 May 2021
Guidance to register the medical devices with MHRA provides details regarding devices that needs to register in the UK. In this guidance, manufacturer, device, and importer attributes has been updated. The excel list of these attributes has been added in the guidance.
US FDA
FDA Issues Draft Guidance on Early Clinical Studies for Certain Medical Devices to Improve Glycemic Control for Type 2 Diabetes| 19 May 2021
The FDA issues a draft guidance for early clinical studies for devices that are intended to therapeutically improve glycemic control for type 2 diabetes. This draft recommends clinical study parameters like study design, sample size, study duration, follow up schedule, effectiveness endpoints etc. The draft guidance is issued for comment purposes only.
FDA Announces Draft Guidance to Help Increase Transparency, Assist Reporting and Timely Completion for Certain Medical Device Studies after FDA Approval or Clearance| 26 May 2021
FDA monitors additional data on certain devices even after FDA clearance or market approval of the device. FDA publishes two guidance documents for post market surveillance of moderate (class II) and high-risk devices (class III). This will assist manufacturers of a device to follow the FDA requirement for ongoing data collection when device is marketed. The draft guidance is issued for comment purposes only.
HEALTH CANADA
Priority COVID-19 test applications: Notice to manufacturers, importers and distributors| 7 May 2021
Health Canada issues a notice to manufacturers, importers and distributors of testing devices related to COVID-19. The following testing technologies are on the highest priority for application evaluation by health Canada: • self-testing devices • point-of-care antigen or molecular testing devices that use nasal swab or saliva samples for use in symptomatic and asymptomatic populations administered by trained operators • point-of-care antigen tests that do not use only nasopharyngeal (NP) swab samples, or may be used in asymptomatic people or may be administered by trained operators • point-of-care molecular tests that do not use only NP swab samples, or may be used in asymptomatic people or may be administered by trained operators • tests designed to address emerging variants • tests that offer new or unique advantages compared to other tests of the same type
• novel diagnostic technologies that may use alternative samples, such as breath, or a different analytical approach
Applications for COVID-19 drug and medical device clinical trials under the interim order: Notice of updated guidance documents| 3 May 2021
Health Canada has updated 2 guidance documents to support Interim Order No. 2 Respecting Clinical Trials for Medical Devices and Drugs Relating to COVID-19. The guidance documents for medical devices is for applicants who wish to sell or import COVID-19 medical device for purpose of clinical trial or to conduct clinical trial for a COVID-19 medical devices authorized under Interim order No.2. Applicant must submit the application in the form of form to the minister.
Interim Order No. 2 respecting clinical trials for medical devices and drugs relating to COVID-19: Notice| 3 May 2021
Ministry of health approves Interim order No. 2 to support the optional pathway to facilitate clinical trials for COVID-19 drugs and medical devices. It addresses any clinical trial submissions that are outstanding when IO No. 1 expires and authorization for drugs and devices issued under IO No. 1. IO No. 2 will assist in expanding the range of applicants who will be able to apply for medical device clinical trial authorization.
Switzerland Swissmedic
New regulations applicable to medical devices as of 26 May 2021 | 26 May 2021
As the Institutional Agreement is missing (InstA) is missing, the EU has not updated the Mutual Recognition Agreement (MRA). No barrier free access will be allowed to Switzerland manufacturers to the EU internal market. Switzerland now has been downgraded to the third country for EU. Swiss manufactures have stricter requirements when exporting their medical devices to the EU. Manufacturers outside the Swiss needs to appoint Swiss Authorised representative in Switzerland. They will need to follow. Medical Device Ordinance MedDO.
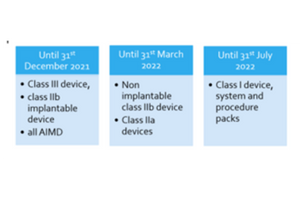
The deadline to appoint a Swiss AR is as follows:
Swiss AR deadline
AUSTRALIA TGA
Medical devices reforms: Reclassification of certain medical devices| 12 May 2021
The TGA is considering consulting regarding the proposed medical device classification for storage solutions for human cells, tissues and organs, and IVF media to simplify issues raised in submissions, prior to considering regulatory amendments for this category of devices. Their aim is to harmonize the classification rule prioritizing the patient safety.
Guidance for Declaration of Conformity| 20 May 2021
TSA released a guidance for declaration of conformity procedures for Class I non-sterile, non-measuring medical devices, Class 1 in vitro diagnostic (IVD) devices, Class I Medical Device (Export Only) and Class 1 IVD Medical Device (Export Only), and Class I Systems and Procedure Packs. This guidance provides information to help manufacturers in completion of corresponding declaration of conformity and assist sponsors to confirm that all the required documentation by manufacturer is complete.
Including IVD medical devices in the ARTG| 19 May 2021
TSA has published a guidance on inclusion of IVD medical devices in the ARTG. ARTG is a register of therapeutic goods that are imported to, supplied in or exported from Australia. Sponsor in the Australia can make an application to include the IVD in the ARTG if the devices comply with the essential principles and appropriate conformity assessment procedure has been carried out. The guidance also explains the different pathways to include the various IVDs like IVD for export, Class 1 IVD, IVD other than Class 1.
Medical device inclusion process| 19 May 2021
TSA publishes the guidance for medical device inclusion process. This process is to include the medical devices in in the Australian Register of Therapeutic Goods (ARTG). This guidance provides detailed information on steps of application to ARTG and applicable guidelines links. According to the current regulation any medical device regulated under The Therapeutic Goods Act 1989, The Therapeutic Goods (Medical Devices) Regulations 2002, The Therapeutic Goods Regulations 1990 needs to be included in the ARTG before importing, supplying, or exporting from Australia.
Therapeutic Goods (Medical Devices-Application Form for Inclusion) Approval 2021| 20 May 2021
TSA approves application forms for Class I, Class IIa, Class IIb, Class III, Class AIMD, Class 1 IVD, Class 2 IVD, Class 3 IVD, Class 4 IVD, Class 4 in house IVD devices to include in ARTG. ARTG is register of therapeutic goods in Australia.
CHINA NPMA
China to strengthen capabilities in drug administration | 10 May 2021
To better protect and promote the health of the public, the General Office of the State Council outlined efforts to enhance the country’s capabilities in drug supervision and management in a circular. Supporting laws and rules will be formulated and revised, refined normative documents and manual to be revised. The circular also emphasized efforts to improve the review and testing system for drugs and medical equipment and cosmetics, intensify the lot release capabilities and strengthen the monitoring system for adverse reactions. It urged building a national platform for drug tracing, applying big data to supervision of drugs, medical equipment, and cosmetics, and using the industrial internet in overseeing vaccines, blood products and special drugs, among others.
Singapore HSA
Consultation for Guidance on the Medical Device Unique Device Identification (UDI) System | 25 May 2021
The Medical Devices Branch (MDB) has published a draft document “Guidance on the Medical Device Unique Device Identification (UDI) System” for comments. This document is intended to provide clarity on the regulatory requirements for Unique Device Identification (UDI) implementation in Singapore and the details on the steps to submit UDI information into the Singapore Medical Device Register (SMDR) and Class a Medical Device Database. The Consultation period for this document is from 25 May 2021 to 30 June 2021.
PAKISTAN DRAP
Draft Guidelines on RAPID ALERTS AND RECALLS ARISING FROM QUALITY DEFECTS GUIDELINES| 27 May 2021
The DRAP has issued a draft guidance document applicable for both human and veterinary products with established quality defects and those reporting with safety and efficacy adverse events. These guidelines are expected to be followed by the licensees (manufacturers, importers, distributors, and retailers. These guidelines aim to explain and standardize the procedure for classification and communications involved in a product recall, to effectively remove therapeutic goods / products from the market that violate requirements and that may cause a health hazard to the consumer/user. The feedback and comments are accepted until due date 17-Jun-2021.
Perform market surveillance of medical devices on UK market and will be able to take decisions over the marketing and supply of device in the UK.
Responsible for designation and monitoring of UK conformity assessment bodies.
UK Approved Bodies
From 1 Jan 2021, the MHRA will be able to designate UK Medical Device Regulation approved bodies to conduct assessments for UKCA marking
Existing UK Notified Bodies with designations under the EU MDD, EU IVDD, EU AIMDD will have their designations rolled over automatically, without having to undergo a new designation process.
Manufacturers of Class I medical devices and general IVDs will be able to self-declare their conformity against Part II and Part IV of the UK MDR 2002 (in the form in which they exist on 1 January 2021), before affixing a UKCA mark and placing the device on the Great Britain market.
Class I medical devices that are sterile or have a measuring function will still require approval from an Approved Body in order to be affixed with the UKCA mark and placed on the Great Britain market.
UK Approved Notified Bodies
Update from NB’s
Manufacturers
Authorised Representatives
GB based AR not recognised in EU from 01 Jan 2021
Manufacturer based outside the EU has to appoint a AR in the EU to sell in the EU and UK based AR will not be accepted.
Importer & Distributor
If Importer is not the UK Responsible Person, the importer will be required to inform the relevant UK Responsible Person of their intention to import a device.
UK Responsible Person will be required to provide the MHRA with a list of device importers.
Existing obligations around storage, transportation and checking device labels for the CE marking or UKCA marking will continue to apply.
The importer’s name and address will not need to be present on the label unless the importer or distributor are acting as the UK Responsible Person.
Registrations in Great Britain
After the transition period, any medical device, IVD or custom-made device will need to be registered with the MHRA before being placed on the Great Britain market.
In Great Britain, devices must conform to the UK Medical Device Regulation 2002, the EU MDR (until 30 June 2023), or the EU IVDR (until 30 June 2023) in order to be registered with the MHRA.
This will apply to devices of all classes.
Where any changes to registrations are made, a £100 standard fee will apply per application.
Registration Timeline – UK
Registration Data
• Copy of Declaration of Conformity
• E-copy of technical documentation
• Device Certificate as granted by the Notified Body
• Quality Management Certificate
• Database information Manufacturing details
• Letter of Designation
• Contact persons and list of importers
• Database information Device Details
• Database information Product Details
• Catalog/Reference (REF)
• Unique Device Identification
Responsibility of UK – Responsible Person
Device registration with MHRA before placing device in UK market.
Ensure declaration of conformity and technical documentation is present & appropriate conformity assessment procedure has been carried out
Keep a copy of the technical documentation, declaration of conformity, relevant certificate, including any amendments and supplements for inspection by the MHRA.
Provide the MHRA with all the information and documentation on request
Cooperate with the MHRA on any preventive or corrective action taken to eliminate or, if that is not possible, mitigate the risks posed by devices.
Immediately inform the manufacturer about complaints and reports about suspected incidents related to a device for which they have been designated.
Terminate the legal relationship with the manufacturer if the manufacturer acts contrary to its obligations under these Regulations and inform the MHRA and, if applicable, the relevant notified body of that termination.
Labelling Requirements
As of 1 January 2021, medical devices placed on the Great Britain market will need to have either a UKCA mark or a CE mark, depending on which legislation the device has been certified under.
Where relevant, the number of the Notified Body or Approved Body will also need to appear on the label.
If you already have a valid CE mark on your device, you will not be required to re-label the device with a UKCA mark until 1 July 2023 for placement on the Great Britain market.
Devices have both marks present in labelling prior to 1 July 2023 and dual marking will be accepted on the UK market after 1 st July 2023.
From 1 Jan 2021 the name and address of the UK RP , where applicable, will need to be included on the product labelling where the UKCA mark has been fixed.
Post-market surveillance and vigilance
Once a medical device has been placed on the UK market, the manufacturer will continue to be required to submit vigilance reports to the MHRA when certain incidents occur in the UK Medical Device Regulation that involve their device.
They must also continue to take appropriate safety action when required. The manufacturer will need to ensure their device meets appropriate standards of safety and performance for as long as it is in use.