
According to European Union Medical Device Regulation (EU MDR) the term “Vigilance” is the identification, reporting and trending of serious incidents and the conduct of safety-related corrective actions (Market surveillance and vigilance).
An ‘incident’ as per article 2(64) MDR is any malfunction or deterioration in the characteristics or performance of a device made available on the market, including use-error due to ergonomic features, as well as any inadequacy in the information supplied by the manufacturer and any undesirable side-effect.
A ‘serious incident’ as per Article 2(65) MDR is an incident as outlined in Article 2(64) MDR that has in addition, either led to or has the potential to lead to significant health or public health outcomes.
In essence, serious incidents refer to a particular category of incidents that have caused, have the potential to cause, or may cause death or severe harm to a patient, user, or any other person.
These incidents can also pose a significant threat to public health. As per Article 87(1) to (5) of the MDR, it is the responsibility of the manufacturer to report such serious incidents to the appropriate regulatory authority.
Any incident which meets all three basic reporting criteria A to C listed below is considered a serious incident and must be reported to the relevant competent authority:
- An incident has occurred
Examples:
- A malfunction or deterioration in the characteristics or performance of the device, e.g. a device that fails or is losing its ability to achieve its intended purpose when used as indicated in the information supplied by the manufacturer
- A deterioration in the characteristics of the device that is related to manufacturing errors, e.g. sterilization process failures
- A use error due to ergonomic features, e.g. a use error caused by a mismatch between the user interface and the physical or medical condition of the intended user
- Any inadequacy in the information supplied by the manufacturer, e.g. insufficient information on how to maintain, adjust or calibrate the device in the instructions for use
- Unclear instructions in the labelling or the manufacturer’s instructions for use
- Undesirable side-effects e.g. allergic skin reactions such as allergy to nickel or wound therapies
- Incident directly or indirectly led, might have led or might lead to any of the outcomes of a serious incident such as death of a patient, user or other person; temporary or permanent serious deterioration of a patient’s, user’s or other person’s state of health; or a serious public health threat. Read more on the concept of vigilance in EU MDR on our website.
Examples of serious public health threats:
- Contagious illnesses, such as HIV, Ebola, Zika virus, SARS and COVID-19
- Events involving high risk of exposure to a disease (e.g., cancer) after use of a medical device, which affects a specific patient population (diabetics, cardiac patients, etc.) or a vulnerable population (children, pregnant women, etc.)
- Exposure to toxic compounds with a potentially negative/harmful effect on humans
- Widespread distribution of falsified or incorrectly labelled devices leading to multiple serious incidents, e.g. distribution of non-sterile devices labelled as sterile
- Cyberattack related to life supporting or life-saving devices
- Causal relationship between the serious incident and the manufacturer’s device has been established or is reasonably possible or suspected.
User, Use error and Abnormal use of a medical device
According to Article 2(37) of the MDR, a “User” refers to any healthcare institution, healthcare professional, or lay person (such as a caregiver or patient) who uses a device, as well as those who install or maintain the device. The term “operator” may also be used to refer to the user of a device.
A “use error” occurs when the actions or inactions of the user while using the device result in a different outcome than intended by the manufacturer or expected by the user.
This can be due to various reasons such as lack of attention, memory lapses, mistakes during device use, or insufficient knowledge or understanding of device use.
Such use errors are not considered incidents, but if they are caused by ergonomic features of a device, they are considered incidents and must be reported under Article 87(1) of the MDR in case of serious incidents.
“Abnormal use” refers to the intentional violation of the intended use of a device. This may involve deliberate acts or omissions that deviate from the normal use of the device and cannot be controlled by the manufacturer.
For example, when a doctor uses a device for an indication that is not specified in the manufacturer’s instructions for use.
“Use-error due to ergonomic features” refers to use errors caused by the physical features of a device that are designed to ensure safe, effective, and efficient interaction between the user and the device.
These errors may occur when there is a mismatch between the device features and the user profile or the environment in which the device is used.
Ergonomic features can be described as the physical features of a device that are designed to facilitate and ensure that the interaction between the user and the device is safe, effective and efficient.
Ergonomic features include components such as measurement and monitoring features, display scales, alarms, software menus, and other factors related to the user interface.
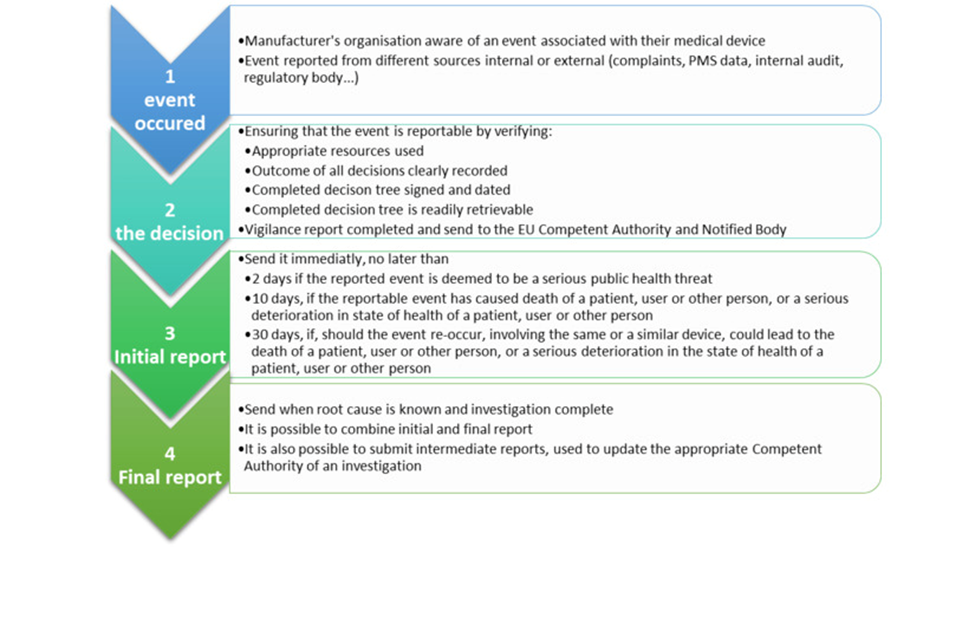
Reporting under MDR
The MDR mandates that the timeline for reporting serious incidents should be based on the severity of the incident. These reporting periods are counted as calendar days.
Typically, the reporting period begins the day after the manufacturer becomes aware of a potentially serious incident. The awareness date, which is considered day 0, is when the manufacturer first receives information about the incident, not after any investigation has been conducted.
The manufacturer’s awareness date is determined as the day when any employee or representative of the manufacturer’s organization first receives information, such as a complaint, about the potentially serious incident.
This can be illustrated with an example:
On June 1, 2022, a manufacturer receives a complaint but decides that it does not meet the criteria for a serious incident, so they do not submit a Manufacturer Incident Report (MIR) to the relevant authority.
However, on July 1, 2022, the manufacturer receives more information and reviews it, and now determines that the complaint is indeed a serious incident. The manufacturer must thus submit a MIR no later than July 16, 2022.

In the MIR, the manufacturer should provide the relevant dates in the following two fields:
- ‘Manufacturer awareness date’ – in this field, the initial awareness date of the incident should be inserted by the manufacturer.
- ‘General comments’ – in this field, the manufacturer should insert the date in which it received the information that determined that the incident is reportable
A ‘field safety corrective action (FSCA)’ is a corrective action taken by a manufacturer for technical or medical reasons to either prevent or reduce the risk of a serious incident, which is associated with a device that is made available on the market.
A Field Safety Corrective Action (FSCA) can involve various actions such as returning the device to the supplier or recalling it, exchanging the device, modifying it, retrofitting it with the manufacturer’s modification or design change, destroying it, giving advice on the device’s use, recommending inspections/examinations of the device by the user, making changes to the device’s software/firmware, and updating the device’s version (for example, rolling it back to an earlier version).
Example of an FSCA conducted by a manufacturer:
As a part of monitoring their products after they have been released to the market, the manufacturer has found a consistent issue with one of their devices.
If this issue could result in a serious problem and the affected devices are still being sold, the manufacturer is required to initiate a FSCA (Field Safety Corrective Action) to prevent or minimize the risk of such incidents.
This FSCA may involve making permanent or temporary changes to the device’s labeling or instructions for use, or recalling all of the affected devices from the market. The manufacturer must promptly inform the relevant authorities in the Member States where the FSCA is being carried out.
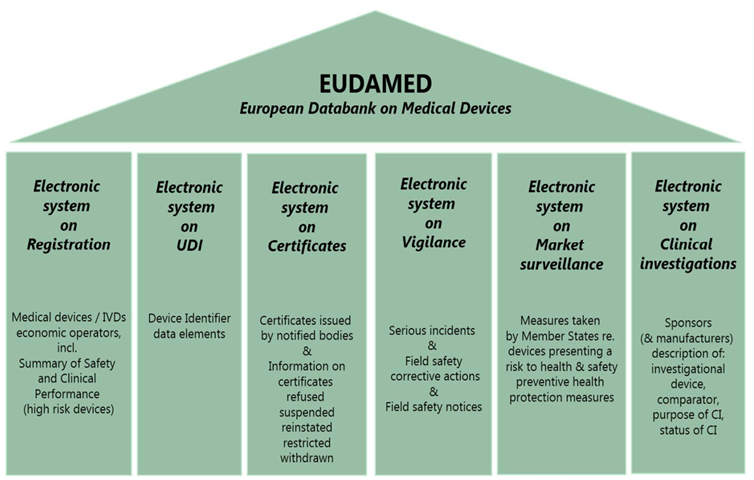
Vigilance Reporting in Eudamed
Eudamed is a recently established database by the European Union for medical devices, designed to gather all necessary information about devices that have been made available in the Union market.
The details of the database can be found in Article 33 MDR, while the rules regarding vigilance are described in Article 92 MDR. The Medical Device Coordination Group (MDCG) guidance provides suggestions on temporary measures that can be used to comply with certain MDR requirements related to Eudamed and information sharing.
These alternatives enable Member States and other stakeholders to fulfill their obligations under the MDR until the complete operation of the database.
Periodic Summary Report
A “Periodic summary report” (PSR) is another way for manufacturers to report serious incidents involving the same device or device type in a consolidated manner.
This reporting method is used when the manufacturer agrees with the relevant national competent authority that coordinates periodic summary reporting.
The PSR is based on certain criteria, which includes situations where the root cause of the incidents has been determined, a Field Safety Corrective Action (FSCA) has been implemented, or where the serious incidents are commonly known and documented.
A ‘common and well documented serious incident’ as referenced in Article 87(9) MDR, must be clearly identified in the manufacturer’s risk analysis and should have led to incident reports, which have been assessed by the manufacturer and the relevant competent authority.
FAQs
Who is responsible for vigilance activities related to medical devices?
The manufacturer of a medical device is responsible for vigilance activities related to that device. This includes monitoring the device’s performance, investigating any incidents or complaints related to the device, and reporting any adverse events to the relevant authorities.
What is a trend report?
A trend report is a summary of adverse event data related to a particular medical device or group of devices. It is used to identify patterns or trends in adverse events and to assess the potential risks associated with the device.
What are the basic reporting criteria for a serious incident?
· An incident (Article 2(64) MDR) has occurred
· The incident directly or indirectly led, might have led or might lead to any of the outcomes of a serious incident (Article 2(65) MDR)
· A causal relationship between the serious incident and the manufacturer’s device has been established, is reasonably possible or suspected