
Before entering the US market, one of the most critical steps for any medical device manufacturer is determining the correct FDA medical device classification.
Your device classification directly impacts: -> Regulatory pathway (510(k), De Novo, PMA) -> Documentation requirements -> Cost and timelines -> Compliance obligations
Misclassification can lead to: -> Delayed approvals -> Rejections -> Increased regulatory burden
In this guide, we will explain FDA device classification in detail, including real examples and how it affects your approval strategy.
Ready to Streamline Your Regulatory Compliance?
Join hundreds of companies who trust OMC Medical for their regulatory needs. Get expert guidance and ensure compliance across all markets.
Call Now +44 208 066 7260What is FDA Medical Device Classification?
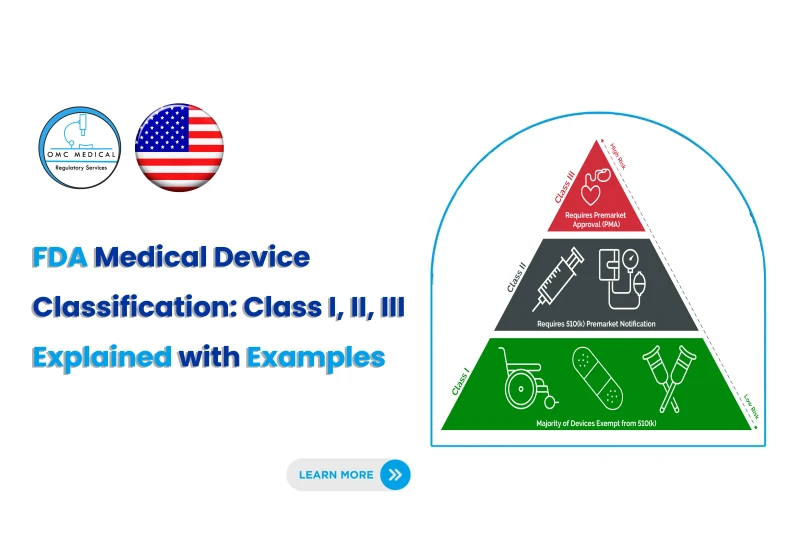
The FDA classifies medical devices into three categories based on risk level: -> Class I – Low risk -> Class II – Moderate risk -> Class III – High risk
👉 The classification determines the level of regulatory control required to ensure safety and effectiveness.
Why Classification is Important
Correct classification is the foundation of your regulatory strategy.
It determines: -> Whether you need a 510(k) submission process -> If your device qualifies for De Novo -> Whether a full PMA is required -> Applicable FDA user fees (as explained in your MDUFA guide)
👉 In short, classification defines your entire FDA approval journey.
Class I Medical Devices (Low Risk)
Overview
Class I devices are considered low risk and are subject to the least regulatory control.
Most Class I devices are: -> Exempt from 510(k) -> Subject to general controls only
Examples of Class I Devices Manual stethoscopes Bandages Surgical gloves (some cases require 510(k)) Handheld surgical instruments
Regulatory Requirements Establishment registration Device listing Labeling compliance Quality system requirements (basic level aligned with Current Good Manufacturing Practices (CGMP))
👉 Even if exempt from 510(k), compliance is still mandatory.
Class II Medical Devices (Moderate Risk)
Overview
Class II devices carry moderate risk and require additional regulatory controls.
Most Class II devices require: 510(k) clearance Demonstration of substantial equivalence
Examples of Class II Devices Infusion pumps Powered wheelchairs Pregnancy test kits Surgical drapes
Regulatory Requirements 510(k) submission Performance testing Labeling requirements Post-market surveillance
👉 A strong strategy for selecting a predicate device is critical—covered in your FDA Predicate Device Selection Strategy for 510(k) blog.
Class III Medical Devices (High Risk)
Overview
Class III devices are high-risk and usually support or sustain life or are implanted.
They require the highest level of regulatory scrutiny.
Regulatory Requirements Premarket Approval (PMA) Clinical trial data Extensive safety and effectiveness evidence
👉 These devices involve higher FDA user fees (MDUFA) and longer approval timelines.
How to Determine Your Device Classification
Determining classification requires a structured approach:
1. Identify Intended Use
What is the purpose of your device?
2. Analyze Risk Level
Consider potential harm to patients.
3. Search FDA Database
Find similar devices and classifications.
4. Check Product Code
Each device is assigned a specific FDA product code.
👉 If no similar device exists, your product may follow the De Novo pathway, as explained in your 510(k) vs De Novo guide.
FDA Classification and Approval Pathways
Here’s how classification connects to approval:
👉 Some exceptions apply, especially for innovative devices.
Common Classification Mistakes
Avoid these frequent errors:
❌ Misinterpreting Intended Use
Leads to incorrect classification.
❌ Selecting Wrong Product Code
Results in submission rejection.
❌ Ignoring Predicate Devices
Affects 510(k) eligibility.
❌ Underestimating Risk
May lead to regulatory non-compliance.
👉 These mistakes often delay approval and increase costs.
How Classification Impacts Cost and Timeline
Your classification directly affects:
Class I
Lowest cost
Fastest market entry
Class II
Moderate cost
Medium timeline
Class III
Highest cost
Longest approval time
👉 Understanding this helps in planning your regulatory budget and strategy.
How OMC Medical Can Help
Accurate classification is critical for successful FDA approval.
OMC Medical provides: Device classification assessment Regulatory pathway strategy 510(k), De Novo, and PMA support Documentation and compliance assistance End-to-end FDA consulting
👉 Our experts ensure your device is correctly classified and aligned with the fastest approval pathway.
Conclusion
FDA medical device classification is the foundation of your US regulatory strategy. Class I → Low risk, minimal requirements Class II → Moderate risk, 510(k) pathway Class III → High risk, PMA required
Getting classification right from the start helps: Avoid delays Reduce costs Ensure compliance
A well-planned approach ensures smooth and successful entry into the US market.
FAQs
Q1. How do I know my device classification?
You can check the FDA database or consult regulatory experts.
Q2. Are all Class I devices exempt from 510(k)?
No, some still require submission.
Q3. Can classification change?
Yes, FDA may reclassify devices over time.
Q4. What if my device has no classification?
It may qualify for the De Novo pathway.





