

A medical device’s intended use and inherent risks must be considered when determining its MDR classification. Class I devices pose less risk to patients and end users, as under the previous MDD. The new Regulation EU MDR 2017/745 has added extended rules, leading some devices to fall under Class IIa, IIb, or even III.
This document is intended to guide Class I medical device manufacturers (excluding custom-made devices) that sell products bearing their name or trademark on the Union market to comply with MDR requirements. This guidance should also apply when an importer, distributor, or any other legal entity takes on the obligations owed by manufacturers.
The class I manufacturer must also hire a Notified Body (NB) to perform a conformity evaluation if medical devices are to be provided sterile, have measuring functions, or are reusable surgical instruments. For the NB to be qualified to conduct such a conformity assessment, the specific medical device in question must come within the purview of the Notified Body’s designation.
Large and mid-sized medical device manufacturers must also properly designate at least one individual in charge of regulatory compliance, but micro and small companies can function better by having a regulatory expert.
Ready to Streamline Your Regulatory Compliance?
Join hundreds of companies who trust OMC Medical for their regulatory needs. Get expert guidance and ensure compliance across all markets.
Call Now +44 208 066 7260It is also important to designate an authorised representative residing within the EU if the manufacturer is registered outside of the EU. A foreign manufacturer must specifically issue a mandate outlining the representative’s duties and authority. It is also crucial to note that not all responsibilities associated with the medical equipment can be assigned.
The manufacturer must adequately preserve all the records required to verify compliance with the relevant requirements so they may be made available to the regulatory body upon request.
Steps to placing Class I medical devices on the market
Manufacturers must ensure compliance with each of the following requirements if they plan to commercialise Class I medical devices. Please be aware that some of the outlined requirements are interconnected and can be completed in a different sequence than that which is shown.
The manufacturer will carry out a gap analysis for Class I devices that have already been released onto the market in compliance with the MDD to ensure that all of the below-listed requirements were satisfied at the time the MDR was applied.
The whole procedure includes a set of mandatory steps, including:
Step 0: Integrate MDR in the Quality Management System (QMS)
The manufacturer’s QMS should be appropriately linked with the standards outlined in the MDR. To ensure compliance with the following requirements, this will enable the correct assessment or decision to be taken and the appropriate documented evidence to be produced.
Step 1: Confirm product as a medical device
The intended purpose and the product’s mode of action are reviewed as per Article 2(1) of the MDR to ensure that the product qualifies as a medical device. If a product is assigned multiple intended purposes, it will qualify as a medical device only if all the intended purposes are covered in Article 2(1).
In the case of products identified as more than one category, the relevant legislation requirements will have to be followed. Despite not being medical devices in and of themselves, accessories to medical devices are subject to MDR regulations and qualify as devices under the MDR definition.
However, the MDR does not include accessories to devices covered by it as per Annex XVI of the MDR.
Step 2: Confirm product as a Class I medical device
The product has to be confirmed if it can be classified as a class I device as per Annex VIII of the MDR. Products previously classified as class I under MDD should be reviewed according to the classification rules of MDR to confirm if reclassification is required.
The MDD guideline cannot be used for devices that were moved from Class I to higher risk classes by the adoption of the MDR. The intended use of the device and any inherent risks related to the period of use, the part of the body, whether it is active or not, and whether it is invasive or non-invasive will determine how the classification standards are applied.
The classification rule with the highest class should be applied if the device in question falls under the purview of more than one classification rule due to its attributes.
Step 3: Procedures before placing on the market
a) Meet the General Safety and Performance Requirements (GSPR)
The devices will adhere to the general safety and performance standards outlined in Annex I of the MDR, considering the intended purposes that their manufacturers had provided.
The Risk management system is a continuous iterative process throughout the entire lifecycle of the product, established by the manufacturer that will enable the identification and analysis of the risks related to each device, the estimation and assessment of the risks associated with those risks, the elimination or control of residual risks, and the evaluation of the adopted measures based on the data gathered from the PMS system.
When a harmonised standard is in place, but a manufacturer chooses to use another reference, implementing that reference should, at the very least, ensure the same level of performance and safety.
Compliance with the relevant harmonised standards will give rise to a presumption that the MDR’s requirements, or portions of them, are also complied with.
If there are available standard specifications, the manufacturer must adhere to them unless they can adequately demonstrate that they have chosen a solution at least as effective and safe. The clinical evaluation processes, risk management, and PMS must be interdependent and updated regularly.
b) Conduct Clinical Evaluation
As part of the MDR’s technical documentation requirements, all devices—regardless of risk classification—require a clinical evaluation. The level of clinical evidence required to show compliance with the pertinent general safety and performance requirements listed in Annex I, which is obtained by considering the characteristics of the device and its intended purpose, shall be specified, and justified by the manufacturer.
Manufacturers must prepare, carry out, and record a clinical evaluation in compliance with Article 61 and Part A of Annex XIV to accomplish this.
Conformity to Annex I requirements can only be assumed when the following items are aligned with each other.
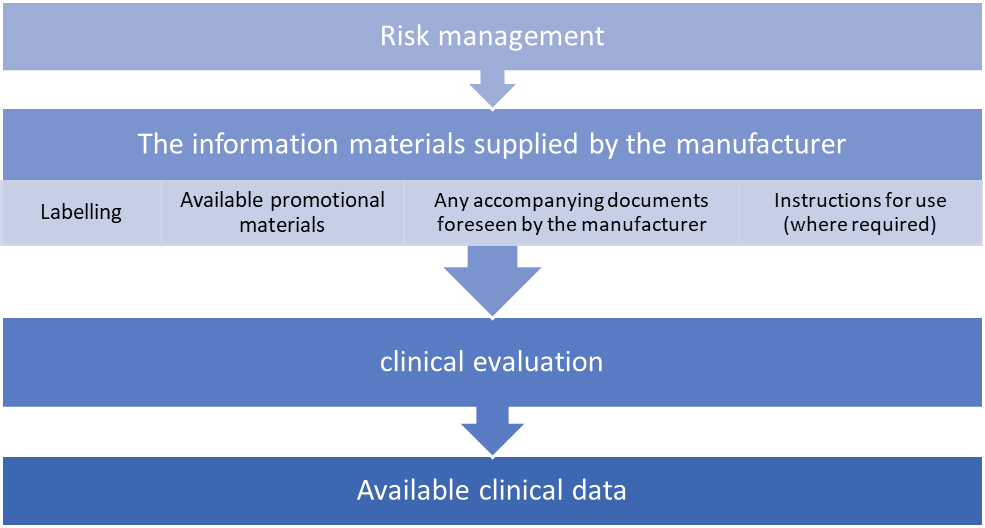
Consideration of available alternative treatment options, the incorporation of clinical data and the acceptability of the benefit-risk ratio are required for carrying out a clinical evaluation.
Additional clinical data will be acquired or developed by clinical investigations if the currently available clinical data are insufficient to establish compliance with the MDR.
If the clinical data currently available for a device that is currently certified under Directive 93/42/EC is insufficient to show compliance with MDR, then post-market clinical follow-up studies of the device may be used to gather more clinical data.
Even data from the general post-market follow-up may occasionally be enough to close the difference.
a) Prepare technical documentation
The manufacturer is responsible for creating and maintaining the technical documentation to prove that their products adhere to the MDR’s technical specifications. Under Annex II and III, this technical documentation must be prepared before the EU declaration of compliance is drawn.
The manufacturer will develop and provide the technical documentation and, if applicable, it’s summary in a way that is unambiguous, clear, well-organised, and easy to search.
The manufacturer shall provide the CA, the authorised representative (where applicable), and NB with access to the technical documents (when applicable).
After reviewing the general safety and performance standards, as well as the pertinent technical provisions of the MDR, the technical documentation will be developed.
b) Request Notified Body involvement
Class I devices do not require the engagement of an NB for MDR compliance. But the manufacturer must follow the guidelines outlined in Chapters I and III of Annex IX or Part A of Annex XI of MDR when the product is a sterile equipment, measuring instrument, or reusable surgical tools.
For the relevant codes and corresponding types of devices as established by Regulation (EU) 2017/2185, manufacturers may select any NB designated in accordance with the MDR.
c) Prepare Instructions for Use and Labelling
Any safety and performance information required for a device’s safe usage and identification of the device should be provided along with the device by the manufacturer and/or the authorised representative, considering the possible users’ training and knowledge.
This data is included on the label, in the device packing, and in the instructions for use. Class I devices do not need instructions for use if they may be operated effectively and securely without them, deviating from the general rule.
Since Class Ir devices will need instructions for reprocessing (cleaning and sterilisation), an exception is most likely proposed.
Labelling and instructions for use must be written in accordance with national language regulations. There will be versions of the labelling and IFU in the technical documentation (in each pertinent national language).
The requirements regarding the information to be supplied with the device will be found in Annex I, Chapter III (23).
Step 4: Check compliance with general obligations for manufacturers
The manufacturer shall ensure that the general obligations for manufacturers outlined in Article 10 are met before releasing a device for sale.
Implementing a suitable QMS that will most effectively assure compliance with the MDR, such as through an internal audit, will receive special consideration. The QMS shall be documented, applied, maintained, updated regularly, and developed continuously, and it will at least include the following elements:
1. A strategy for regulatory compliance
2. Identification of applicable general safety and performance requirements and exploration of options to address those requirements
3. Responsibility of the management
4. Resource management, including selection and control of suppliers and sub-contractors
5. Risk management
6. Clinical evaluation, including post-market clinical follow-up (PMCF)
7. Product realisation, including planning, design, development, production and service provision
8. Verification of the UDI assignments
9. Setting up, implementation and maintenance of a post-market surveillance system
10. Handling communication with competent authorities, notified bodies, other economic operators, customers and/or other stakeholders
11. Processes for reporting serious incidents and field safety corrective actions in the context of vigilance
12. Management of corrective and preventive actions and verification of their effectiveness
13. Processes for monitoring and measurement of output, data analysis and product improvement.
According to applicable Union and national law, either natural or legal persons may seek compensation for harm brought on by a defective equipment.
In order to protect themselves against potential responsibility under Directive 85/374/EEC, manufacturers must take precautions that are commensurate to the risk class, type of device, and size of the business. These steps must not compromise further protective measures under national legislation.
Step 5: Draw-up the EU Declaration of Conformity
The process by which the manufacturer, who complies with the standards established by Article 52(7), certifies that the devices in question comply with the requirements of the MDR that apply to them is referred to in Article 19 as the EU declaration of conformity.
The CA will have access to the declaration of compliance, which must include, at a minimum, all of the data referred to in Annex IV. The manufacturer will regularly update the EU declaration of conformity and translate it into the official language(s) required by the Member States where the product is sold.
Suppose a device is subject to additional Union laws in addition to the MDR that also call for an EU declaration of conformity. In that case, the manufacturers will create a single EU declaration of conformity that refers to all applicable Union laws.
By creating the EU declaration of conformity, the manufacturer accepts liability for the device’s regulatory compliance with all relevant Union legislation.
For class Ir, Im, and Is devices, the manufacturer must obtain an EC certificate from NB per Annex IX, Chapters I and III, or Annex XI, Part A before applying a CE mark.
Step 6: Affix the CE Marking
All Class I medical devices on the market must display the CE conformity label, which must be visible, readable, and permanent. It may be applied to the item or its sterile packaging. The CE marking must be applied to the package in cases where such affixing is impossible or unwarranted due to the nature of the device. The CE certification must be visible on all sales packaging and the instructions for use.
Placing the CE mark on a medical device signifies that it complies with all applicable safety and performance standards and is approved for marketing in the EU. The CE marking will be accompanied by the identification number of the relevant NB in the case of Class I medical devices placed on the market in a sterile condition, devices with measurement functions, and/or reusable surgical tools.
Affixing marks that could lead third parties astray about the significance of the CE marking is banned. Other extra markings may be applied to the product, the packaging, or the usage instructions, but they must not obscure or obstruct the CE marking.
The CE marking format will follow Annex V requirements. The minimum dimensions required for the CE mark may not apply to very small devices.
Step 7: Registration of devices and manufacturers in Eudamed
A streamlined approach might be used for medical devices that were previously put on the market per the Directives, provided that the manufacturer evaluates the gap and ensures that all regulations are properly followed.
A Class I medical device manufacturer must register the product with Eudamed before putting it on the market. Suppose the information listed in Section 1 of Part A of Annex VI has not already been registered in accordance with Article 31. In that case, the manufacturer must submit it to the electronic system in order to register the device. The information referred to in Section 1 of Part A of Annex VI will be given to that electronic system before applying to the NB in situations where the conformity assessment method necessitates the engagement of an NB in accordance with Article 52.
Following the CA’s validation of the manufacturer’s data in Eudamed, the manufacturer will receive an SRN from the aforementioned electronic system. To fulfil its duties under Article 29, the manufacturer will use the SRN when submitting an application to an NB for a conformity assessment and to gain access to Eudamed.
The registration of a device in Eudamed by the manufacturer includes:
1. Assigning a UDI-DI (with a Basic UDI-DI) as described in Part C of Annex VI to the device in accordance with the issuing entity’s policies mentioned in Article 27(2) and adding the UDI-DI (with a Basic UDI-DI) to the UDI database along with the other core data elements pertaining to that device mentioned in Part B of Annex VI.
2. Entering the data referred to in Section 2 of Part A of Annex VI, excluding Section 2.2 thereof, or, if previously provided, validating the data, and then keeping the data current up to date.
This document summarises the information presented above and outlines how Class I medical devices should be introduced to the EU market. The guidance’s scope also includes reusable surgical instruments (Class Ir), sterile medical devices (Class Is), and devices with measurement capabilities. These medical devices all need a Notified Body to be involved in pre-marketing procedures.





