Medical device apps are increasingly growing these days. MHRA has issued new guidance on the stand-alone software medical devices, including apps. This guidance is a crucial document for manufacturers and users of such medical devices.
In the UK, medical devices are subject to UKCA marking. The UKCA marking is no exception to software medical devices.
Software Medical Devices or not
Classifying software or a mobile app as a medical device can be challenging. If the software or app has a well-defined intended medical purpose, it is essential to mark CE or UKCA on the product.
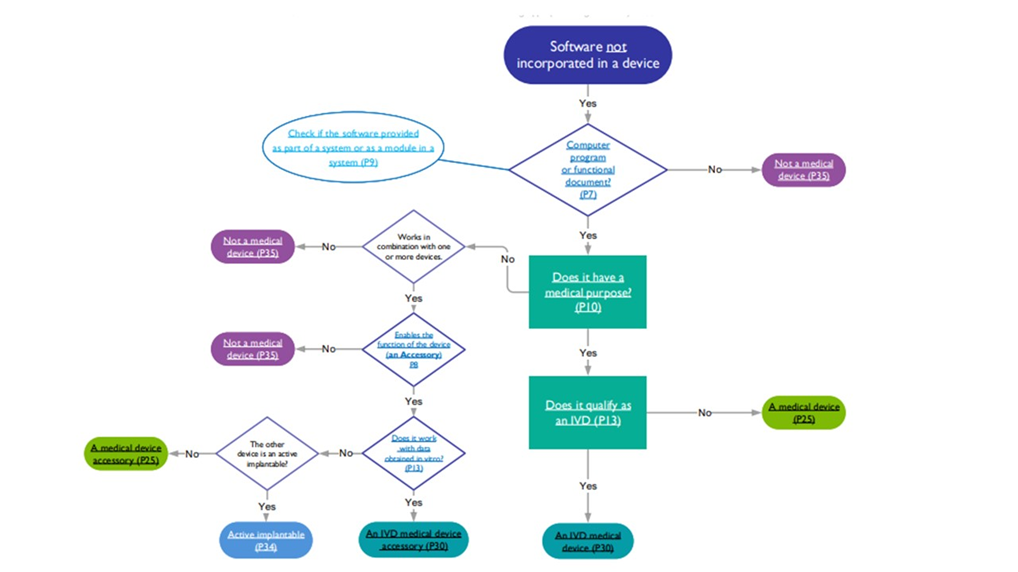
This ensures that the device conforms to the requirements of the regulation of the EU and UK and is safe for use. The flow chart provided below helps determine if the software is a medical device, in vitro diagnostic device, active implantable, or accessory.
Flowchart on the classification of software into medical device Source: MHRA Software flowchart
Intended purpose
A medical device is defined by the intended purpose on the device labelling, Instructions for use and any promotional materials, including brochures.
Depending on the intended purpose, the device can be classified as a device with a medical purpose if it:
Prevents disease
Diagnoses a disease, injury or handicap
Monitoring a disease, injury or handicap
Treats or alleviation of a disease, injury or handicap
Compensates an injury or handicap
Investigates replaces or modifies anatomy or physiological process
Controls conception
A software device is considered to have a medical purpose if it has one of the following features and looks into in vitro data:
Concerning a physiological or pathological state
Concerning a congenital abnormality
To determine the safety and compatibility with potential recipients
To monitor therapeutic measures
Software Medical device classification and essential requirements
Rule 9 includes active therapeutic devices intended to administer or exchange energy
Rule 10 includes active devices intended for the diagnosis
Rule 12 includes all other active class I devices
Rule 14 includes devices used for contraception or the prevention of the transmission of sexually transmitted diseases
One of the essential requirements for software apps is for the benefit to outweigh any risks. Essential requirements also state the risks of ergonomic features and the intended use environment.
Manufacturers of such devices must ensure that the user interface must be consistent, and graphics and text must be clear and legible. The software or app must be designed with safety in mind.
In addition, the clinical evaluation following Annex X of the UK MDR must be done. A similar set of requirements applies to IVDs.
Labelling requirements
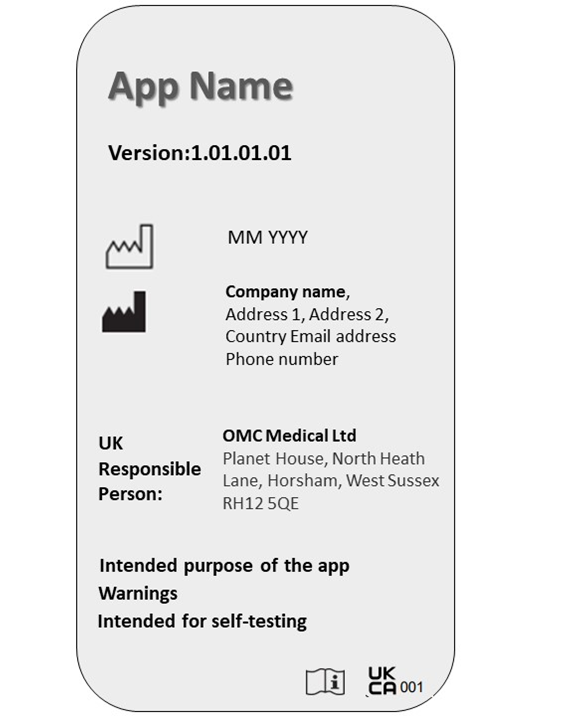
The software labelling must be clear and visible to the user. Manufacturers must ensure the app meets relevant requirements and displays UKCA marking on the landing page itself.
The following particulars must be present on the software label:
App name
Version number
Date of manufacture
Manufacturer name and address
UKRP name and address
Purpose of software
Warnings and precautions
UKCA or CE marking
Labelling for an app with medical purpose
Examples of Software with a medical purpose
Software with a medical purpose may be devices that:
Provide information for the calculation of drug dose using IVD data such as blood
Enables therapeutic drug monitoring
Monitor blood glucose levels
Provides medical conditions based on input user data
Indicate potential developing disease based on the entered data
Automate the pathway for treatment for an individual
Enables people with visual or hearing disabilities to read or listen by magnifying text or amplifying sounds
Software or app without an intended medical use would include:
General apps for recording patient images which later require the diagnosis of a clinician
Apps that give general recommendations instead of user-specific advise
Software that is intended to record heart rates, such as fitness or sports apps
Apps that remind patients of drug intake
Apps to treat non-medical conditions
Software that provides tips or advice
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.
European Medical Device Nomenclature (EMDN) – Questions and Answers | 15 June 2021
The European Commission (MDCG) published a question and answers article for European Medical Device Nomenclature (EMDN). European Medical Device Nomenclature (EMDN) supports the functioning of the European database (EUDAMED). EMDN will be used for the registration of medical devices in EUDAMED by manufacturers. It explains the term EMDN, how EMDN is created, its key principles, the structure of EMDN.
MDCG 2021-13 Questions and answers on obligations and related rules for the registration in EUDAMED of actors other than manufacturers, authorized representatives, and importers subject to the obligations of Article 31 MDR and Article 28 IVDR | 23 June 2021
The document aimed at addressing questions relating to the registration in EUDAMED of actors other than manufacturers, authorized representatives, and importers subject to the obligations of Article 31 of Regulation (EU) 2017/745 on medical devices (MDR) and/or Article 28 of Regulation (EU) 2017/746 on in vitro diagnostic medical devices (IVDR). It also clarifies the cases where an Actor ID is issued instead of an SRN.
Joint implementation and preparedness plan for Regulation (EU) 2017/746 on in vitro diagnostic medical devices (IVDR) | 7 June 2021
European commission has issued a plan for implementation of in vitro diagnostic medical devices regulation (EU) 2017/746. This plan serves as a living document to monitor their implementation. The status and timelines of the items will be updated to reflect the progress of the work.
UK MHRA
Operation Pangea: Officers from the Medicines and Healthcare products Regulatory Agency (MHRA) have seized millions of illegally traded medicines and medical devices | 8 June 2021
In a week of action coordinated by Interpol, this year’s ‘Operation Pangea’ ran from 18 to 25 May and saw over 100 countries joining forces to seize non-compliant medical products and to identify and remove thousands of illegally operating websites and URLs offering medicines and devices. The operation also involved coordinating the arrests of several suspected organized criminals. The MHRA will be following the week of action with a detailed analysis of the global results to create a better understanding of current and emerging threats. This work includes the identification of ‘hotspot’ exporting countries, favoured high-risk medicines being traded on the black market, and the ever-evolving business models of criminals worldwide seeking to take advantage of the public.
US FDA
Clinical Outcome Assessments (COAs) in Medical Device Decision Making | 21 June 2021
A clinical outcome assessment (COA) describes or reflects how a person feels, functions, or survives and can be reported by a health care provider or a non-clinical observer, through performance of an activity or task or by the patient. For regulatory purposes, high-quality information from COAs can provide valuable evidence for benefit-risk assessments and can be used in medical device labeling to communicate the effect of a treatment on patient symptoms and functioning. COAs may also be used to help measure the safety of the device and measure how well the device performs in treating or diagnosing the condition.
HEALTH CANADA
Off-label advertising and sale of rapid antigen tests under workplace screening program: Interim Enforcement approach | 15 June 2021
There are currently various technologies to detect SARS CoV-2, the virus that causes COVID-19. While some rapid antigen detection tests (RADTs) have been approved for people without symptoms,
most RADTs are indicated for use on people with symptoms and are to be conducted by laboratory personnel, healthcare professionals or trained operators. Health Canada has authorized several RADTs under two interim orders:
1. interim order No. 1 for importing and selling medical devices (March 18, 2020, to March 1, 2021)
2. interim order No. 2 for importing and selling medical devices (enacted March 1, 2021)
The interim enforcement discretion will be in effect until December 31, 2021. The exception is if :
1. Post-market monitoring identifies new risks or
2. There is no longer a need to apply this discretion based on public health status.
AUSTRALIA TGA
Custom made medical devices | 30 June 2021
On 25 February 2021, a new framework for regulating personalized medical devices commenced. The framework includes a new definition for custom-made medical devices. The impact of the new definition is most devices previously supplied under the exemption for custom-made medical devices no longer meet the definition of a custom-made medical device and will need to be included in the Australian Register of Therapeutic Goods (ARTG).
Medical device reforms: Conformity Assessment Bodies | 30 June 2021
Conformity assessment is the systematic and ongoing examination of evidence and the application of procedures to ensure a medical device complies with the essential principles for medical devices. Evidence that a device has undergone an appropriate conformity assessment procedure must be held by the manufacturer before a device can be included in the Australian Register of Therapeutic Goods (ARTG). Previously, Australian medical device manufacturers could only apply for a conformity assessment certificate from either the TGA or an overseas Notified Body. To provide additional flexibility and timeliness, the Australian Government agreed to regulatory changes that allow other Australian corporations that demonstrate appropriate experience and competence to undertake conformity assessment of medical devices. An Australian CAB must demonstrate they can perform product assessments and quality management system audits under the Australian conformity assessment body framework. The TGA remains responsible for including medical devices in the Australian Register of Therapeutic Goods (ARTG).
Regulatory changes for custom-made medical devices | 21 June 2021
On 25 February 2021, a new regulatory framework commenced changing the way the Therapeutic Goods Administration (TGA) regulates medical devices that are designed, manufactured, assembled, or adapted to meet the needs of an individual. The changes are collectively referred to as the personalized medical devices framework (the Framework). The Framework introduced the following:
On 25 February 2021, a new regulatory framework commenced changing the way the Therapeutic Goods Administration (TGA) regulates medical devices that are designed, manufactured, assembled, or adapted to meet the needs of an individual. The changes are collectively referred to as the personalized medical devices framework (the Framework). The Framework introduced the following:
New definitions for types of personalized medical devices, greatly reducing the number of devices that can be supplied under the custom-made medical device exemption.
New conditions of exemption for custom-made medical devices, in the form of requirements to: • submit an annual report detailing all custom-made medical devices supplied in the previous financial year. • Allow the TGA to inspect production facilities. • retain documentation about custom-made medical devices for 5 years (for Non implantable devices) or 15 years (for implantable devices). • Provide information about each custom-made medical device to the intended recipient.
The new concept of a Medical Device Production System (MDPS) which, once fully implemented, will provide options to healthcare providers wishing to produce personalized devices for treating their patients and updates to the classification rule for medical devices that record diagnostic images to include a broader range of technology now used for the purposes of recording patient anatomy for diagnosis and investigation, including anatomical models.
SINGAPORE HSA
Mean applicant screening response time | 23 June 2021
HSA strives to complete the screening of the new and major variation applications in the shortest possible time. For the new and major variation applications accepted within the period of 01 Oct 2020 to 31 Mar 2021, the mean screening time taken by HSA was 31.9 Working Days (WD) for New Drug Application (NDA), 33.6 WD for Generic Drug Application (GDA) and 19.2 WD for Major Application Variation (MAV) applications, respectively.
Below are bi-annual updates of the mean applicant response time for the new and major variation applications:
Mean Applicant Response Time
Period
NDA
GDA
MAV
01 Oct 2020 to 31 Mar 2021
Number of applications
62
109
79
Mean Applicant Response Time (WD)
33.2
40.2
17.0
PAKISTAN DRAP
Notification regarding amendments in form-2 of the Medical Devices Rules,2017 | 7 June 2021
The Drug Regulatory Authority of Pakistan (DRAP) on recommendation of the Medical Devices Board, made the following amendments in Form-2 sub-rule (3) of rule 63 of the Medical Devices Rules, 2017: The entries in column (2) at sub-serial number (iv) and (viii) of serial number 2 shall be omitted and remaining entries shall be renumbered accordingly.
SWITZERLAND SWISSMEDIC
Information from Swissmedic about MedDO | 19 June 2021
If the manufacturer of a medical device does not have its registered place of business in Switzerland, its products may only be placed on the market once an authorized representative domiciled in Switzerland has been appointed (Art. 51 para. 1 MedDO). This also applies to manufacturers with their registered place of business in the EU. The transitional periods defined in Art. 104a MedDO apply to the authorized representative.
Perform market surveillance of medical devices on UK market and will be able to take decisions over the marketing and supply of device in the UK.
Responsible for designation and monitoring of UK conformity assessment bodies.
UK Approved Bodies
From 1 Jan 2021, the MHRA will be able to designate UK Medical Device Regulation approved bodies to conduct assessments for UKCA marking
Existing UK Notified Bodies with designations under the EU MDD, EU IVDD, EU AIMDD will have their designations rolled over automatically, without having to undergo a new designation process.
Manufacturers of Class I medical devices and general IVDs will be able to self-declare their conformity against Part II and Part IV of the UK MDR 2002 (in the form in which they exist on 1 January 2021), before affixing a UKCA mark and placing the device on the Great Britain market.
Class I medical devices that are sterile or have a measuring function will still require approval from an Approved Body in order to be affixed with the UKCA mark and placed on the Great Britain market.
UK Approved Notified Bodies
Update from NB’s
Manufacturers
Authorised Representatives
GB based AR not recognised in EU from 01 Jan 2021
Manufacturer based outside the EU has to appoint a AR in the EU to sell in the EU and UK based AR will not be accepted.
Importer & Distributor
If Importer is not the UK Responsible Person, the importer will be required to inform the relevant UK Responsible Person of their intention to import a device.
UK Responsible Person will be required to provide the MHRA with a list of device importers.
Existing obligations around storage, transportation and checking device labels for the CE marking or UKCA marking will continue to apply.
The importer’s name and address will not need to be present on the label unless the importer or distributor are acting as the UK Responsible Person.
Registrations in Great Britain
After the transition period, any medical device, IVD or custom-made device will need to be registered with the MHRA before being placed on the Great Britain market.
In Great Britain, devices must conform to the UK Medical Device Regulation 2002, the EU MDR (until 30 June 2023), or the EU IVDR (until 30 June 2023) in order to be registered with the MHRA.
This will apply to devices of all classes.
Where any changes to registrations are made, a £100 standard fee will apply per application.
Registration Timeline – UK
Registration Data
• Copy of Declaration of Conformity
• E-copy of technical documentation
• Device Certificate as granted by the Notified Body
• Quality Management Certificate
• Database information Manufacturing details
• Letter of Designation
• Contact persons and list of importers
• Database information Device Details
• Database information Product Details
• Catalog/Reference (REF)
• Unique Device Identification
Responsibility of UK – Responsible Person
Device registration with MHRA before placing device in UK market.
Ensure declaration of conformity and technical documentation is present & appropriate conformity assessment procedure has been carried out
Keep a copy of the technical documentation, declaration of conformity, relevant certificate, including any amendments and supplements for inspection by the MHRA.
Provide the MHRA with all the information and documentation on request
Cooperate with the MHRA on any preventive or corrective action taken to eliminate or, if that is not possible, mitigate the risks posed by devices.
Immediately inform the manufacturer about complaints and reports about suspected incidents related to a device for which they have been designated.
Terminate the legal relationship with the manufacturer if the manufacturer acts contrary to its obligations under these Regulations and inform the MHRA and, if applicable, the relevant notified body of that termination.
Labelling Requirements
As of 1 January 2021, medical devices placed on the Great Britain market will need to have either a UKCA mark or a CE mark, depending on which legislation the device has been certified under.
Where relevant, the number of the Notified Body or Approved Body will also need to appear on the label.
If you already have a valid CE mark on your device, you will not be required to re-label the device with a UKCA mark until 1 July 2023 for placement on the Great Britain market.
Devices have both marks present in labelling prior to 1 July 2023 and dual marking will be accepted on the UK market after 1 st July 2023.
From 1 Jan 2021 the name and address of the UK RP , where applicable, will need to be included on the product labelling where the UKCA mark has been fixed.
Post-market surveillance and vigilance
Once a medical device has been placed on the UK market, the manufacturer will continue to be required to submit vigilance reports to the MHRA when certain incidents occur in the UK Medical Device Regulation that involve their device.
They must also continue to take appropriate safety action when required. The manufacturer will need to ensure their device meets appropriate standards of safety and performance for as long as it is in use.