ISO – International Organization for Standardization, is the international, non-governmental body for drafting and establishing technical and non-technical standards.
These standards are developed by different committees within the International Organization for Standardization. Having around 165 member states, with one representative from each, International Organization for Standardization is a global entity catering to the needs of industry requirements.
Are ISO standards important?
The International Organization for Standardization medical device standards are the Bible for many countries, especially ones which do not have predefined regulations or processes.
In addition to general standards, ISO also publishes product-specific guidance such as for Implants, Orthopedic, Medical Electric Equipment, and many more.
Global International Organization for Standardization Requirements
In Europe, the European Commission has the Medical Device Regulation MDR 2017/745 and In-vitro Diagnostic Device Regulation IVDR 2017/746.
These regulations provide a detailed framework for introducing a medical device in the European market. However, in addition to that, certain International Organization for Standardization may also be referred to for ensuring a better-quality product.
Some of the many popularly used standards include:
ISO 14971:2019 Medical Devices – Application of Risk Management to medical devices
ISO 15223-1:2021 Medical devices – Symbols to be used with information to be supplied by the manufacturer – Part 1: General requirements
IEC 60601-2-83 Medical electrical equipment – Part 2-83: Particular requirements for the basic safety and essential performance of home light therapy equipment
IEC 60601-1 Medical electrical equipment – Part 1: General requirements for basic safety and essential performance
The European Commission also has Harmonized Standards, developed by European Standards Organization CEN, CENELEC, or ETSI, per the international standards.
It provides a list of the applicable harmonized standards for enhanced product safety and quality.
In the USA, the US Food and Drug Administration (FDA) has a Code of Federal Regulations (CFR) and Guidance.
CFRs are legally binding. Manufacturers must comply with the requirements of CFR
The guidance provides Agency’s thinking on regulatory issues. They are NOT legally binding
In addition to these, the FDA also accepts certain recognized consensus standards from different organizations such as International Organization for Standardization, CLSI, ANSI, IEC, CEN, etc.
These standards may be used to justify a Declaration of Conformity for a product. The widely accepted medical device International Organization for Standardization are, but are not limited to:
ISO 10993 – Biological Evaluation for Medical Devices
ISO 14160 – Sterilization of Healthcare Products
ISO 11737 – Sterilization of Medical Devices
In Canada, the Standards Council of Canada (SCC) is the International Organization for Standardization member body. Similar to the US FDA, the Therapeutic Products Directorate (TPD) of Health Canada periodically releases a list of acceptable international or national standards for medical devices.
Manufacturers can use these recognized standards in conjunction with the Health Canada’s Medical Devices Regulations (SOR-98/282) and the Guidance Documents, to prove product conformity and safe use in the market.
China‘s National Medical Products Administration (NMPA) is developing indigenous standards that more closely align with those of ISO. Biocompatibility testing is one avenue where the scope and requirements for China are more than that of the US/EU.
Hence, NMPA has developed various biocompatibility testing standards which are to be used in addition to the International Organization for Standardization standard.
For the rest of the world’s medical device industry,
India encourages International Organization for Standardization certification for all its industries. The medical sector must be International Organization for Standardization 13485 compliant while the pharmaceutical sector must be ISO 9001 compliant for Quality Management Systems, in addition to other relevant and applicable International Organization for Standardization.
Japan’s The Japanese Industrial Standards Committee (JISC) is an International Organization for Standardization member body. The regulatory authority, Pharmaceutical and Medical Device Agency (PMDA) revised its Ordinance No. 169 in 2021 to closely align with the International Organization for Standardization 13485:2016 standard. The transition period is 3 years and must comply by March 25, 2024
For the Korean regulatory authority, aligning the requirements of Korean Good Manufacturing Practice (GMP) to that of International Organization for Standardization 13485:2016 is believed to be a step closer to entering the Medical Device Single Audit Program (MDSAP)
Russia’s Federal Service for Surveillance in Healthcare (Roszdravnadzor) is known to accept International Organization for Standardization 13485:2016 certification. Information on acceptance of other International Organization for Standardization cannot be confirmed. It does not accept market approvals in the US, EU, or other countries as a reference for market authorization in Russia
Australia’s Standard Australia is a member of the International Organization for Standardization, IEC, and ICSID. It strongly encourages the use of international standards, except where their use is ineffective or inappropriate and does not develop any national Australian standard for which there is already an international standard in existence. In 2019, TGA published Therapeutic Goods (Conformity Assessment Standard for Quality Management Systems) Order 2019which provides a list of applicable conformity assessment standards.
Brazil’s ANVISA accepts Good Manufacturing Practices (GMP) along with the International Organization for Standardization 13485
FAQs
Can QMS be established solely based on ISO standards?
For countries that do not have their own QMS regulations, the ISO standard can be used as a reference. For countries with established local regulations, and that accepts ISO, both ISO standard and local/national regulations must be considered.
Are ISO standards freely available?
No. ISO standards are available for purchase from the ISO official website. However, they do have FREE read-only formats available.
Comparing ISO standards to local regulations, which one takes precedence?
The local or national regulation always takes precedence over the ISO standard.
Can the manufacturer use an older version of an ISO standard for compliance?
No. Manufacturers must make sure they comply with the active or most recent version of the ISO standard. This is not restricted just to ISO standards but applies to National regulations too. Manufacturers must keep their QMS up to date with the latest requirements of the industry. The ideal way to be updated is to refer to the latest version of any Standard or Regulation.
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.
The FDA 510(K) Pre-market notification submission as per 21 CFR 807 Subpart E is to be adopted by manufacturers to receive FDA clearance to market medical devices or for commercial distribution in the U.S.
This review is done by FDA’s Centre for Devices and Radiological Health (CDRH). A 510(K) submission allows medical devices to be “FDA Cleared” and not FDA Approved.
The route to 510(K) must be carefully investigated by the manufacturer through a step-by-step process which allows determining if the regulatory pathway chosen for the Medical Device’s FDA access is in the right direction.
Step 1: Decision Criteria Checklist
The assessment checklist helps the manufacturer to arrive at a decision if they are eligible or fall under the rules to submit an FDA 510(K) application.
Criteria
510(K) Submission required?
Are you a domestic manufacturer willing to commercially distribute your product in the U.S?
ü
Are you developing specifications for a Finished device and you have an external firm/contractor who manufactures the device based on your specifications?
ü
Are you a repacker or re-labeller who makes significant changes to the device operations such as changing label contents/warnings/safety signs / operating conditions to the original device label prior to sale to the market?
ü
Are you a foreign manufacturer?
ü
Are you making changes to an existing 510(K) cleared finished device where the changes could significantly affect the device’s safety and effectiveness?
ü
Are you making changes to the intended use of the medical device?
ü
Do you manufacture accessories for a medical device that are sold directly to the end-user as replaceable/serviceable parts?
ü
Do you sell unfinished devices or components to another firm that places the Finished device for sale using your components in their device?
û
Are you willing to introduce your finished devices for clinical trials only to the market? (this means you are only subjecting your device for clinical trials and not commercially distributing them)
û
Are you acting the role of a distributor for a domestically manufactured device by affixing only labels indicating “distributor” or “manufacturer” details?
û
Are you an “Importer” who is willing to import a foreign manufactured device and that device has already been 510(K) cleared?
û
Is your device either Class I or Class II and falls under the Medical Device Exemptions 510(K) and GMP requirements of the FDA?
û
Step 2: Device Classification
The next step toward submission is to verify how the medical device is classified under the FDA classification regulations.
There are different generic types of devices identified by the FDA and placed under 3 categories of regulatory classes based on the risk posed by the medical device and the level of controls necessary for the safety and effectiveness of the device.
Class I Devices Low-Risk Devices – General Controls
With Exemptions
Without Exemptions
The above states that certain class I low-risk devices are exempted from the “General Controls”.
Class II Devices Moderately Risk Devices – General Controls and Special Controls
With Exemptions
Without Exemptions
The above states that certain class II devices are exempted while others fall under the provisions of “General and Special Controls”.
Class III Devices High-Risk Devices – General Controls and Pre-market Approvals
Pre-Market Approval
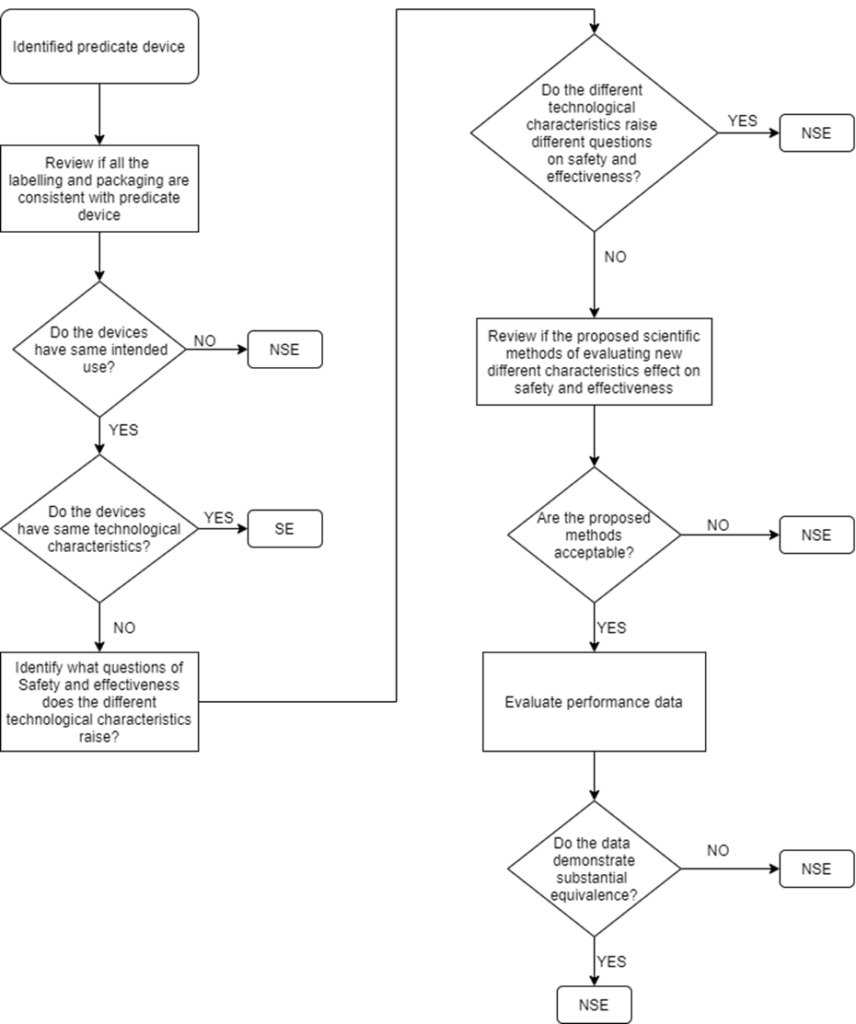
Step 3: Determine if your device is Substantially Equivalent to a Predicate Device
510(K) submission is applicable only for devices that can claim Substantial Equivalence (SE) to a predicate device. Below flowchart is an illustration that helps to clearly understand the decision route:
“SE” – Substantial Equivalent
“NSE” – Non-Substantial Equivalent
Multiple Predicate Devices
In certain cases, the manufacturer may identify more than one predicate device i.e., multiple predicates. In such cases, the primary predicate refers to the one that is most similar to the below factors:
Intended Use
Indications for use
Technological characteristics
The manufacturer is recommended to identify the most appropriate primary predicate device with a well-supported decision document.
Supporting Documents to Claim Substantial Equivalence
The following are required by the manufacturer but not limited to, while demonstrating the most appropriate predicate device and that the new device to be submitted for 510(K) is a substantial equivalent to a predicate device.
Intended Use
Indications for Use
Technological Characteristics (similarities, differences and whether the differences pose different questions on safety and effectiveness)
Performance data to support substantial equivalence (biocompatibility testing, Electrical safety and Electromagnetic Compatibility, Software verification and validation testing, mechanical testing, clinical study, animal study, if applicable)
A declaration of conformity to recognised standards applicable to the medical device
Refer to the Section 807.92 content and format of a 510(K) summary.
Step 4: Determine the Type of 510(K) Submission
Within the 510(K) applications, there are 3 categories of submissions as discussed below
SL.
Submissions
Type of 510(K) applicable
1
To introduce a new medical device into the market which has a predicate device available
Traditional 510(K)
2
If a manufacturer introduces changes introduced to the device that is already existing in the market and has obtained a 510(K) clearance
Special 510(K)
3
If submission relies on FDA guidance documents voluntary consensus standard demonstration of compliance with special controls for the device type
Abbreviated 510(K)
Step 5: The 510(K) Submission Process
Step 5.1: FORM FDA 3601 Medical Device User Fee Cover Sheet
Take a printed copy of this user fee cover sheet. This could be the first page of the 510(K).
Step 5.2: FORM FDA 3514 CDRH PREMARKET REVIEW SUBMISSION COVER SHEET
Download form FDA 3514 pdf. This form captures detailed information required for the different types of submissions.
A cover letter and/or the FDA Form 3514 should follow the User fee cover sheet. If FDA Form 3514 is not affixed, then the cover letter should contain all the elements relevant to the submission contained in Form 3514. This will expedite the processing time.
Step 5.3: 510(K) Submission Acknowledgement receipt by FDA
If a valid eCopy and a proper user fee has been paid Acknowledgment Letter get received from DCC through email. If the proper fee and a valid eCopy are submitted by the holder, then the holder receives an acknowledgement letter from the DCC through an email.
The following are identified by the Acknowledgement Letter:
Receipt’s date (the date that FDA received the 510(k) submission, an eCopy and the proper user fee payment);
The receipt’s date (this is the day that 510(k) submission was received by FDA, valid eCopy, and proper user fee payment); and 510(k) number
Step 5.4: Document Contents in a 510(K) Submission
Below is the minimum list of contents necessary to be available in 510(K) submission documents.
Cover Letter
Table of Contents
Indications for Use (FDA Form 3881)
510(K) Summary or Statement
Truthful and accurate statement as required by 21 CFR 807.87(l)
Class III Summary and Certification (to be submitted when claiming equivalence to a Class III device) As per 21 CFR 807.94
Financial Certification & Disclosure Statement
Declaration of Conformity and Summary Reports
Proposed Labelling
Sterilization and Shelf Life
Biocompatibility
Device Specifications
Substantial Equivalence Comparison
Software
Electromagnetic compatibility and Electrical Safety
Performance Data (Summary on Clinical and Non-Clinical data)
Additional Requirements
Summary
Mode of Submission to FDA – E copies
In section 745A(b)(1) of the Federal Food, Drug, and Cosmetic Act (FD&C Act) (21 U.S.C. 379k-1) FDA is amending its regulations on medical device submissions to remove requirements for paper and multiple copies and replace them with requirements for a single submission in electronic format.
Submissions in electronic format include eCopies, submissions created and submitted on CD, DVD, or flash drive and mailed to FDA, and eSubmissions, submission package produced by an electronic submission template.
Fees, Exemptions and Waivers
Under the user fee system, medical device companies pay fees to the FDA when they register their establishments and list their devices with the agency, whenever they submit an application or a notification to market a new medical device in the U.S. and for certain other types of submissions.
The MDUFA (Medical Device User Fee) website User Fee has information on the current fee charges applicable. Payment must be received and processed at the time or before the date the application is sent.
If the FDA receives an application without full payment of all required fees, the FDA will consider the application incomplete and will not begin its review.
Review Stages
Acceptance Review
This is based on the Refuse to Accept (RTA) policy by the FDA. It is a mechanism adopted by the FDA to provide a quick review of the 510(K) submission.
This stage is only the initial (Acceptance review) stage where the FDA reviewer using separate checklists for each type of submission (traditional, abbreviated and special) reviews the submission and gives a declaration if the submission contents meet the minimum threshold requirements or is placed on RTA hold.
The FDA reviewer evaluates the submission against specific acceptance criteria and informs the submitter within the above timeline on acceptance or indicate the missing element(s) in submission.
In order to enhance the consistency of FDA’s acceptance decisions and to help submitters better understand the types of information FDA needs to conduct a substantive review, this guidance, includes the checklists to clarify the necessary elements and contents of a complete 510(k) submission.
Only if this stage is cleared, the submission gets qualified for the actual Substantive review stage. The reviewer conducting substantive review is the actual Lead reviewer.
Substantive Review
During Substantive Review, the Lead Reviewer conducts a comprehensive review of the 510(k) submission and communicates with the submitter through a Substantive Interaction, which should occur within 60 calendar days of receipt of the 510(k) submission.
Substantive Interaction communication is typically:
an email stating that FDA will proceed to resolve any outstanding deficiencies via Interactive Review; or
an Additional Information (AI) request which places the submission on hold.
Additional Information (AI) request
If the lead reviewer sends an AI request, then it means the submission is placed on hold. The submitter has 180 calendar days to address the queries from the date of additional information request by FDA reviewer.
If the queries are not addressed by the applicant within this time span, then 510(K) submission is deleted from the FDA database and the applicant will need to submit a new 510(K) to pursue the FDA market clearance process.
The submitter must submit the response, with a valid eCopy, to the DCC. The response should:
include the submitter’s name;
list the 510(k) number;
identify the submission as Additional Information (AI) to the 510(k);
list the date of FDA’s request for additional information; and
provide the requested information in an organized manner.
Interactive Review
If the Lead Reviewer chooses to continue with an Interactive Review, this means the Lead Reviewer has determined that any outstanding deficiencies may be adequately addressed within the timeframe set by the Medical Device User Fee Amendment of 2012 (MDUFA III) performance goal for a 510(k) (90 FDA days) and that the submission will not be placed on hold.
The Lead Reviewer communicates with the submitter during the Interactive Review using tools such as:
Email
Telephone Call
During Interactive Review, the Lead Reviewer may request additional information from the submitter, who may either send the information to the Lead Reviewer directly or to the DCC (Document control centre).
Note: During Interactive Review, any information submitted to the DCC must include a valid eCopy.
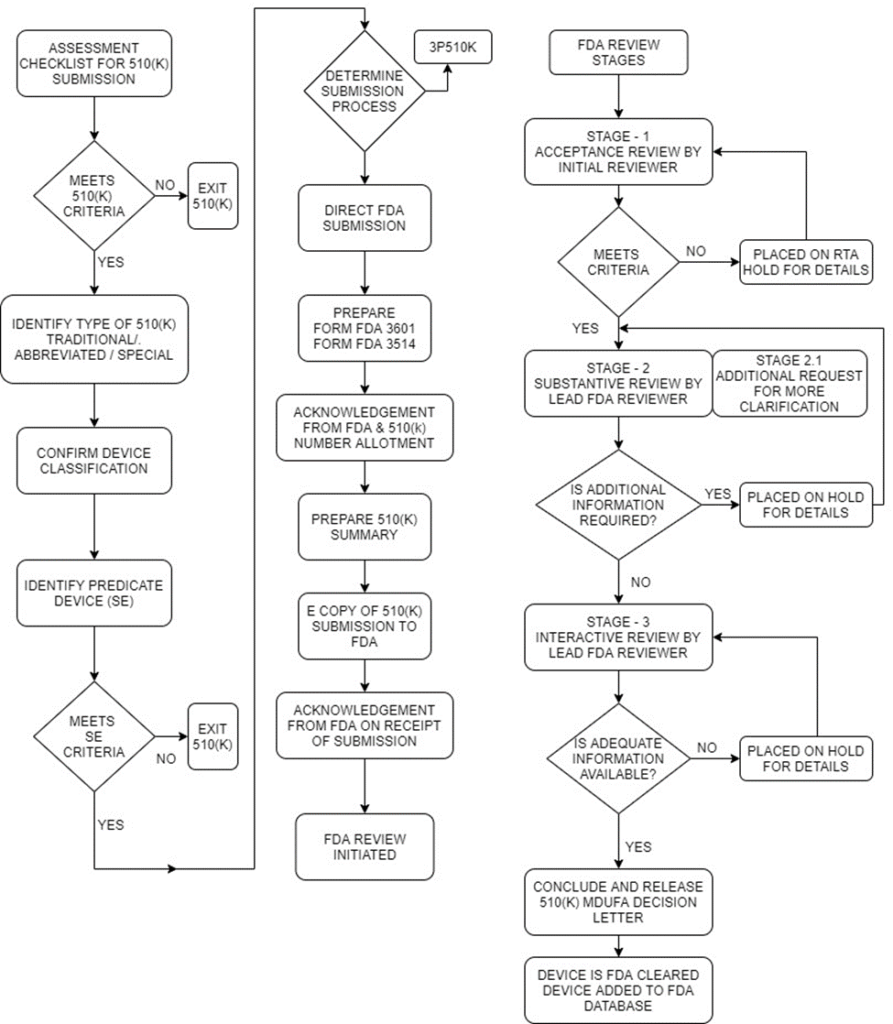
Timeline – An overview of 510(K) Submission
Day:01
FDA receives the 510(K)-application submission
Day:07
FDA sends the acknowledgement letter (or) FDA sends HOLD letter (in case of issues)
Day:15
FDA conducts Acceptance Review and informs the applicant if the application is eligible for substantive review (or)
Places it under RTA Hold
Day:60
FDA conducts Substantiative Review and communicates on the next move towards Interactive Review
Day:90
FDA sends Final MDUFA Decision Letter
Step 6: Final 510(K) Decision Letter
MDUFA Decisions for 510(k) submissions include findings of substantially equivalent (SE) or not substantially equivalent (NSE).
When a decision is made, FDA will issue the decision letter to the submitter by email to the email address provided in the 510(k) cover letter.
A 510(k) that receives an SE decision is considered “cleared.”
FDA adds the cleared 510(k) to the 510(k) database, which is updated weekly.
The IFU and the summary will be sent as attachments to the SE letter. The IFU will not be signed since it is considered an attachment to the SE letter. Therefore, the signature on the SE letter will apply to both the letter and the IFU.
If FDA does not reach an MDUFA decision within 100 FDA days (i.e., 10 days after the MDUFA goal), FDA will issue a Missed MDUFA Communication, which is written feedback to the submitter to be discussed in a meeting or teleconference, including the major outstanding review topic areas or other reasons that are preventing FDA from reaching a final decision, with an estimated date of completion.
510(K) decision-making flow chart
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.
Medical device nomenclatures are those products used to prevent, diagnose, treat, and monitor the many diseases known to humankind. Medical devices and medicines play an equally important role in treating human beings.
To learn more about medical devices, read our article on the definition of a medical device. This article discusses the nomenclature of medical devices and examples of these.
What is the Nomenclature of Medical Devices?
To simply put it, the nomenclature is the naming of a medical device. Although medical devices are classified into different risk classes, they should be named so that it is universally identified. Standardised nomenclature facilitates this easy identification.
A medical device nomenclatures is needed to simplify trade and tracking among the different regulatory authorities, Ministries of Health, and other organisations that regulate medical devices.
A standardised medical device nomenclatures aids in the following aspects:
Grouping and classification of medical devices.
Registration under different regulatory bodies or Ministries of Health
Streamlined procurement and distribution
Grouping of medical devices in various electronic health records and medical device databases
Vigilance reporting, field safety and post-market surveillance
The different medical device nomenclatures available are as follows:
GMDN or Global Medical Device Nomenclatures.
EMDN or European Medical Device Nomenclatures
UMDNS or Universal Medical Device Nomenclatures System
Other nationally developed nomenclature systems
Global Medical Device Nomenclatures (GMDN)
About 10% of countries use Global Medical Device Nomenclatures worldwide. It is a system of internationally accepted descriptors used to identify medical devices. The GMDN Agency manages GMDN codes, a non-profit organisation.
GMDN is a 5-digit code containing the following information:
GMDN Term Name: Anaesthesia ventilator
GMDN Code: 34851
GMDN Definition: A mains electricity (AC-powered) stand-alone, automatic cycling device used to assist and control alveolar ventilation during general anaesthesia and is compatible with inhaled anaesthetic agents. It has fewer functions and is less complex to operate than an intensive care ventilator but adequately meets the patient’s ventilation needs for oxygen (O2) and carbon dioxide (CO2) exchange to maintain normal blood gas concentrations. The device provides a mechanical means to deliver the breathing gas to the patient in a controlled pattern. It is equipped with alarms to warn of changes in respiration or the onset of unsafe operating conditions.
GMDN was introduced for a variety of regulatory purposes. GMDN is based on the ISO 15225: Medical device nomenclature data structure’. Read more about the frequently asked questions about GMDN here.
European Medical Device Nomenclature (EMDN)
European Medical Device Nomenclature or EMDN is introduced due to Article 26 of EU Regulation 2017/745 of Medical devices and Article 23 of EU Regulation 2017/746 of in-vitro diagnostic medical devices.
Like GMDN, it plays a considerable role in device nomenclature and serves various regulatory purposes. One of the primary uses is while registering a medical device in EUDAMEDwhere it is closely linked to UDI-DI.
Structure of EMDN
The European Medical Device Nomenclature is characterised by its alphanumeric structure and is established in a seven-level hierarchical tree where it clusters medical devices into three primary levels:
Categories: the first hierarchical level – alphanumeric.
Groups: the second hierarchical level – 2 numbers indicating group.
Types: the third hierarchical level – a series of numbers 1,2,3,4 and 5.
EMDN was adopted from the Classificazione Nazionale Dispositivi medici (CND) classification. The EMDN can be accessed at the EMDN list. European Medical Device Nomenclature categorises into three primary levels, categories, groups, and types.
A category comprises several groups composed of various kinds of medical devices.
Source: https://webgate.ec.europa.eu/dyna2/emdn/. In the above image of EMDN, ‘A’ is the category, ‘A01’ is Group and ‘A0101’ to ‘A0199’ are the types of medical devices.
Universal Medical Device Nomenclature System (UMDNS)
Universal Medical Device Nomenclature System or UMDNS was developed by the Emergency Care Research Institute (ECRI). Many nations have adopted this standard around the world. UMDNS is used in inventory control, work order control, and regulatory systems applications.
It is a 5-digit code unique code and a term for different types of medical devices. One can find the UMDNS code list here. UMDNS is updated monthly.
CND Nomenclature
CND nomenclature or ‘Classificazione Nazionale Dispositivi medici’ was developed by Italian Ministry of Health. In addition to Italy, it is also used in Portugal and Greece.
The guidance document on CND nomenclature explains the basic principles and structure of CND, which also applies to EMDN as EMDN was adopted from CND nomenclature. Following this, medical devices are clustered into three levels:
Category
Group
Type
FAQs
Is UDI the same as GMDN?
Both Unique Device Identification System (UDI) and GMDN are used in device identification. However, the two have some fundamental differences. UDI is inferior because of its lack of unity. It does not have a structure; therefore, device identification becomes more difficult with UDIs. Nonetheless, it is an effective tool for the traceability of medical devices. FDA utilises UDIs for medical device identification. The user guide to GUDID explains how the UDIs are managed in US FDA’s database, GUDID.
Can EMDN be accessed free of charge?
The EMDN is accessible to all stakeholders- free of charge. Hence, it can be utilised by a non-exhaustive list of stakeholders such as manufacturers, patients, research organisations, practitioners, hospitals, etc. The EMDN can also be downloaded from here.
Is there a guidance document that helps economic operators to map the EMDN information into the forthcoming EUDAMED database?
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.
The FDA introduced current Good Manufacturing practices (cGMP) to maintain a system that provides proper design, monitoring, and control of manufacturing processes and facilities.
The cGMP standards ensure products’ identity, strength, quality, and purity by requiring manufacturers to maintain proper control over their manufacturing processes.
This system helps the manufacturer avoid as many failures and errors as possible. The “C” in cGMP refers to “current,” which means that organisations must employ up-to-date technologies and systems to comply with the rules.
The cGMP is subdivided into the following chapters:
Subpart A-General Provisions (§§ 820.1 – 820.5)
Subpart B – Quality System Requirements (§§ 820.20 – 820.25)
Subpart C – Design Controls (§ 820.30)
Subpart D – Document Controls (§ 820.40)
Subpart E – Purchasing Controls (§ 820.50)
Subpart F – Identification and Traceability (§§ 820.60 – 820.65)
Subpart G – Production and Process Controls (§§ 820.70 – 820.75)
Subpart H – Acceptance Activities (§§ 820.80 – 820.86)
Subpart I – Nonconforming Product (§ 820.90)
Subpart J – Corrective and Preventive Action (§ 820.100)
Subpart K – Labelling and Packaging Control (§§ 820.120 – 820.130)
Subpart L – Handling, Storage, Distribution, and Installation (§§ 820.140 – 820.170)
Subpart M – Records (§§ 820.180 – 820.198)
Subpart N – Servicing (§ 820.200)
Subpart O – Statistical Techniques (§ 820.250)
cGMPs Quality System Requirements
Establishing appropriate policies for maintaining quality requirements and ensuring they are followed in responsibility of the manufacturer. Handling matters such as the responsibility, authority, and interrelation of all personnel, resource allocation, quality plan, quality system procedure, and instructions is the manufacturer’s responsibility.
A representative must be appointed who ensures the quality requirements and periodically reports its performance to the executive management.
Quality audits must be mandatorily performed by personnel who are not directly responsible for the matters under audit and reports of such audits must be documented and shared with executive management. Ensuring availability of trained personnel for each task, and training activities for the same is must.
Design Controls
Every manufacturer, irrespective of any device class should maintain a specific design plan that must include design input requirements and the development process, which should be reviewed and updated periodically.
The manufacturer must maintain procedures to define and document design input and output data and be reviewed regularly. The results of design reviews must be documented in the design history file (DHF).
The manufacturer is also expected to maintain procedures for verification and validation of the design procedure. Design changes and procedures for the changes to product specifications must be part of the manufacturer’s design control plan.
All information listed above must be part of the manufacturer’s Design History File (DHF).
Document Controls
This subpart describes how documents should be verified before distribution.
A responsible for checking document adequacy before circulation with their signature and date of verification is must and should be documented.
Any changes to the documents can be verified by an individual(s) in the same function or organisation unless specifically designated and changes made are to be communicated to the concerned person.
Information such as the description of the change, identification of the affected documents, the signature of the approving individual(s), the approval date, and when the change becomes effective must be part of the change record.
Purchasing Controls
This subpart describes that each manufacturer must maintain a procedure to verify if all purchased or received products conform to the specified requirements.
Manufacturers must evaluate and document their suppliers, contractors, and consultants for all specified requirements, including quality requirements. The type of control over these operators must be defined, and a list of permissible operators should be documented.
It is a must to maintain data with a clear description or reference to specified requirements, including servicing. Purchasing data shall be approved in accordance with Sec 820.40.
Identification and Traceability
This subpart describes identification and traceability requirements.
Manufacturers must establish methods to identify the product during all stages of receipt, production, distribution, and installation to prevent mix-ups. Devices intended for surgical implants or to support or sustain life must have procedures to identify the product with a control number 0f each unit, lot or batch of finished devices. This will help while taking corrective action. Such identification details must be documented in DHR.
Production and Process Controls
This subpart describes how the manufacturer should develop, conduct, control and monitor production processes to ensure that a device conforms to its specifications.
Details of Documented instructions, SOPs, methods that define and control the manner of production, monitoring and control of process parameters, compliance with specified standards, and approval processes criteria for workmanship must be included where process control is required.
Production and process changes must be verified and validated according to Sec 820.75, and changes shall be approved in accordance with Sec 820.40. There must be proper procedure to maintain its working if environmental control is applicable.
Details on personnel, contamination control, buildings, Equipment, Maintenance schedule, Inspection, Adjustment, Manufacturing material and automated processes are mentioned in detail in this section.
Section 820.72 describes inspection, measuring and test equipment. Various aspects from the control of inspection, measuring and testing equipment to its calibration and records are discussed in detail in this section.
Section 820.75 describes process validation. The processes must be verified with a high degree of assurance and approved according to established procedures.
Manufacturers must maintain procedures for monitoring and control of validated processes. Any changes or deviations in the process should be reviewed, evaluated, revalidated if necessary, and appropriately documented.
Acceptance Activities
This subpart describes the process of receiving in-process and finished device acceptance and acceptance status in detail. Finished products shall not be released for distribution until:
(1) The activities required in the DMR are completed (2) the associated data and documentation are reviewed (3) the release is authorised by the signature of a designated individual(s)
(4) the authorisation is dated.
The acceptance record shall include the following details:
(1) The acceptance activities performed (2) the dates acceptance activities are performed (3) the results (4) the signature of the individual(s) conducting the acceptance activities (5) where appropriate, the equipment used. These records shall be part of the DHR
To ensure that the product that has passed the required acceptance activities is distributed, used, or installed, the identification of acceptance status must be maintained throughout the product’s production, packaging, labelling, installation, and service.
Nonconforming Products
This subpart describes the control of nonconforming products and nonconformity review and disposition.
A procedure to identify, document, evaluate, segregate, and disposition a nonconforming product must be maintained by the manufacturer. The evaluation and investigation results must be documented.
The manufacturer must also establish and maintain a procedure for rework, such as retesting and re-evaluating nonconforming products after rework. All these activities must be documented in the DHR (Device History Record).
Corrective and Preventive Action
This subpart describes corrective and preventive actions. Procedures such as analysing, investigating, identifying, verifying, implementing, ensuring correct information, and submitting relevant information for all processes must be established and followed by the manufacturer.
All the nonconforming products must be reported and documented. During the verification process, the manufacturer must ensure that the corrective and preventive action should be effective and should not adversely affect the finished device.
Labelling and Packaging Control
This subpart describes how to establish and maintain procedures to control labelling activities.
Labels affixed to the device must remain legible throughout the manufacturing process and until it is delivered to the end-user.
Manufacturers must establish a procedure for labelling inspection, labelling storage, and labelling operations. During the inspection process, the responsible person must ensure the correct unique device identifier (UDI) or universal product code (UPC), expiration date, control number, storage instructions, handling instructions, and any additional processing instructions present on the label.
A control number as per Sec820.65 for each product must be part of the labelling. This subpart also mentions that the manufacturer must establish a proper procedure for device packaging during different stages of the manufacturing process.
Handling, Storage, Distribution, and Installation
This subpart describes the product’s handling, storage, distribution, and installation.
During storage and handling, the manufacturer must ensure that mix-ups, damage, deterioration, contamination or other adverse effects must not affect the product.
Manufacturers must establish a procedure to authorise the receipt and dispatch the product to storage rooms. Manufacturers must maintain distribution records which should include the following:
(1) The name and address of the initial consignee (2) The identification and quantity of devices shipped (3) The date shipped (4) Any control number(s) used.
Further, Details on Installation procedures to be adopted by the manufacturer are given in detail.
Records
This subpart describes the requirements of record keeping.
The manufacturer is expected to maintain confidentiality if required and a set record retention period for all records as per the nature of the document.
It shall include device specifications, production process, Quality assurance procedures, packaging, labelling, installation, maintenance, and servicing procedures and methods.
The manufacturer must also maintain a Device history record (DHR) which should contain the following information:
(a) The dates of manufacture (b) The quantity manufactured (c) The quantity released for distribution (d) The acceptance records which demonstrate the device is manufactured in accordance with the DMR (e) The primary identification label and labelling used for each production unit (f) Any unique device identifier (UDI) or universal product code (UPC), and any other device identification(s) and control number(s) used
Detailed information on complaint files and Quality system records are also included in this subpart.
Servicing
This subpart describes servicing requirements.
Manufacturers are expected to analyse service reports with appropriate statistical methodology as per Sec 820.100.
Any service report that must be reported to the FDA under part 803 of the CGMP document shall automatically consider the report a complaint and proceed according to Sec 820.198. In general, the service report must include the following details:
(1) The name of the device serviced (2) Any unique device identifier (UDI) or universal product code (UPC) and any other device identification(s) and control number(s) used (3) The date of service (4) The individual(s) servicing the device (5) The service performed (6) The test and inspection data
The last subpart of the CGMP document gives details of Statistical Techniques to be used while following the CGMP guidelines.
FAQs
What are the field safety corrective actions of the FDA?
FDA has a stringent FSCA requirement. Refer to our article to understand the FDA’s field safety corrective action procedures.
Are there exemptions from GMP?
Medical devices published in the Federal Register and codified in 21 CFR 862 to 892 and exempted by FDA classification regulations are exempted from following the GMP requirements. However, manufacturers of finished devices must maintain complaint files (21 CFR 820.198) and general requirements concerning records (21 CFR 820.180). Medical devices manufactured under an investigational device exemption (IDE) are not exempt from design control requirements under 21 CFR 820.30 of the QS regulation.
Is it acceptable for a manufacturer to produce sterile products by aseptic processing to rely solely on ISO standards to qualify the facility?
No, in addition to the ISO standards it is required that the manufacturer follows applicable FDA regulations.
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.
Summary of Safety and Clinical Performance (SSCP) acts as a vital document that allows the public to access information quickly. The information in the SSCP can be sourced entirely from the technical file.
The technical file consists of the Post Market Surveillance (PMS), risk assessment, post-market clinical follow-up (PMCF) plans and reports. The SSCP document is required for high-risk devices only-this includes Class III and all implantable devices.
Manufacturers of custom-made or investigational devices need not produce this document. Implant card together with SSCP enables an efficient system to access device information.
SSCP for medical devices under MDR
Under MDD, the information on medical devices was not easily attainable. Therefore, the end-users of most medical devices were deprived of information regarding even high-risk medical devices.
As a result, medical devices under the directives lacked clarity. The main reason why SSCP is introduced is that MDR, unlike the directives, brings accessibility of information into account.
The SSCP should be made available on the EUDAMED website for easy access. It should be assigned an identifier that remains the same throughout the document’s lifetime. This identifier is not subjected to changes even when the content of this document is revised.
Language and readability requirements
SSCP follows the MDR language requirements like the other technical documents, such as Instructions for Use. All intended users within the EU understand no single language.
Therefore, language translations should be made available to intended users and patients. The SSCP should be translated into the languages accepted in the Member States where the device is intended to be sold.
Healthcare professionals widely understand English. Having an English translation available is essential, even if it is not available in selecting the official languages of each Member State. This enables the access to information that EU MDR strives to achieve.
While preparing SSCP, readability is an essential factor. The manufacturers must bear in mind to produce two sections. One part for intended users/healthcare professionals, and a second part for patients if applicable.
It is recommended to use an appropriate method to confirm that the document is understandable to the member of both categories. Further guidance on the readability, translations and other factors involved in SSCP can be found in the MDCG guidance on Summary of safety and clinical performance.
Sections of Summary of Safety and Clinical Performance
Article 32 of the EU MDR states some sections that are a must. The manufacturer may add further information from the Technical Dossier/File if relevant to the users.
The following sections are mandatory in an SSCP for healthcare professionals:
Details of the device and manufacturer (including UDI details).
Intended use of the device.
Description of the device and its components. Previous variants of the device.
Indications, contraindications, and target demographic.
Details of residual risks, undesirable effects, warnings, and precautions
Risks, undesirable effects, and warnings should be mentioned in SSCP. Other relevant aspects of safety, serious events, and a summary of any field safety corrective action must be included if applicable.
A summary of the Clinical Evaluation of the device.
This section is intended to summarise the clinical evaluation results and the clinical data, the evaluation of undesirable side-effects, and the benefit-risk ratio’s acceptability.
It is an objective and balanced summary of the clinical evaluation results of all the available clinical data related to the device. It should comprise favourable, unfavourable, and inconclusive data.
Conclusions based on evidence and the safety and performance of the device.
Suggested profile and training for users
Applied harmonised standards
All commonly applied specifications and international standards harmonised and adopted monographs should be listed in SSCP.
Revision history
Revision history should contain details such as revision validated by Notified Body (NB) and the language of SSCP validated.
A patient-specific SSCP template follows the same high-level structure as the clinician SSCP but does not include the reference to harmonized standards and common specifications. This decision focused on the information most relevant to patient health.
Validation of SSCP
When the Notified Bodies (NB) have assessed that all the required elements are included in the draft SSCP with the most current version of relevant documents in the TD, the SSCP has been validated by the NB.
SSCP validation may depend on the class of device and the conformity assessment routes chosen. More guidance on validation by NB can be found in the MDCG guidance on SSCP.
Uploading SSCP to EUDAMED
SSCP should be made available online for the users who intend to read the document. It is uploaded in EUDAMED by the Notified Bodies, the only actor managing the SSCPs in EUDAMED.
After each validation process, NB shall upload the updated SSCP by replacing the older version. However, once the ‘master’ SSCP is uploaded, it is up to the manufacturer to upload the translations to EUDAMED.
FAQs
What resources can be used for the SSCP?
The technical files like the design validation report, risk management report, clinical evaluation report (CER), as well as Post-Market Surveillance (PMS) and Post-Market Clinical Follow-up (PMCF) reports.
The Instructions for Use of the device can also be used as information for preparing the SSCP. Please note that SSCP cannot replace the IFU.
How frequently should SSCP be reviewed or updated?
The SSCP should be ideally updated annually. Documents like the PMCF evaluation and periodic safety update reports (PSUR) are updated annually. It is recommended to update SSCP along with the other technical documents.
How can I access the SSCP?
SSCP will be made available with the launch of the EUDAMED database. SSCP is accessible to both healthcare professionals and patients. SSCP has clear information for healthcare professionals with prior knowledge of medical terminologies.
In addition to this, SSCP should also contain a section that patients of different levels of expertise easily understand.
When should a manufacturer of Class III medical devices prepare SSCP?
SSCP should be made available at the time of registration.
Is there a similar requirement under IVDR?
Article 29 of In Vitro Diagnostics Regulation 2017/746 (EU IVDR) describes a requirement like the SSCP: the Summary of Safety and Performance (SSP).
The SSP requirements are almost identical to those of the SSP, replacing ‘clinical’ for ‘performance’ evaluation. Also, it includes a requirement for metrological traceability of assigned values intended for analytes used in IVDs.
For more information on IVDR, read our article on IVDR 2017/746.
Medical Device Reporting (MDR) is one of the FDA’s post-market surveillance techniques for monitoring device performance, detecting potential device-related safety concerns, and contributing to device benefit-risk assessments.
There are mainly two groups of reporters of which the mandatory group includes the manufacturer, importers and device user facilities, and the voluntary group includes healthcare professionals, caregivers, patients, consumers
Once submitted, the FDA reviews the MDR and validates the totality of the given information
The MDR submission by itself cannot act as evidence that the medical device reporting is responsible for a particular adverse event unless the reporter believes that the adverse event has or may have occurred due to the device or the medical device reporting has a role in the mentioned event
What Event Qualifies to be Reported?
A medical device reporting placed in the market contributes to a death or a serious injury
A medical device reporting has malfunctioned and is likely to cause death or a serious injury if the malfunction were to recur
Who can Report an Adverse Event to FDA?
It is considered mandatory for the manufacturers, importers, and device user facilities) of reporters to report any adverse event through form FDA 3500A.
The summary of Requirements for Mandatory Reporting:
Reporter
Description
FDA Form(s)
Report to
When to Report
Manufacturer
30-day reports of deaths, serious injuries, and malfunctions
Within 30 calendar days of becoming aware of an event
5-day reports for an event designated by FDA or an event that requires remedial action to prevent an unreasonable risk of substantial harm to the public health
The voluntary reporting can be done through MedWatch Online reporting Form or by requesting for assistance by calling 1-800-332-1088:
Healthcare professionals are advised to report through form FDA 3500
Patients/Consumers are advised to report through form FDA 3500B
In case of Emergencies, reporting can be done by contacting the FDA Office of Crisis Management, Emergency Operations Centre.
Who is Responsible to carry out Corrective Action for the Reported Event?
For any reportable or reported adverse events, the manufacturer is obliged to carry out a corrective action such that the implemented corrective action and preventive action aims to eliminate any medical device reporting malfunctions related to patient safety.
The manufacturers, importers and the user facilities are primarily responsible for carrying out a corrective action whose goal is to gather data, evaluate it, detect and investigate product and quality issues, and then take suitable and effective corrective action to keep them from happening again.
Top 5 Medical Device Safety Action Plan Objectives
FDA carries out inspections to ensure that the corrective actions undertaken by the manufacturer are well adequate to eliminate any recurring adverse events.
The Key objectives of such inspections are:
To create a medical device patient safety net in the United States of America
To put forward a system to check on the latest regulatory options to implement postmarket mitigations
To guide innovations to make a safer medical device
Bring Improvements in medical device cybersecurity
Integrate the premarket and post-market offices and operations of the Centre for Devices and Radiological Health (CDRH) to promote the implementation of a TPLC (Total product life cycle) approach to device safety
Corrective and Preventive Action (CAPA) Procedure
Every manufacturer is mandated to establish and maintain a CAPA process as per the FDA’s Quality System Regulation (QSR) 21 CFR 820.100.
The process must well contain the requirements on input data sources, decision criteria of non-conformance, investigational procedure, root cause analysis, actions to address the non-conformance, effectiveness verification procedure, implement actions to eliminate recurring problems taking into consideration about the in-house product design changes as well as the impact to existing products in the market.
The FDA requires all these activities to be documented and recorded.
Manufacturers are required to create a report as per 806.10 of any corrective actions implemented on the device and submit it to the FDA
Manufacturers and Importers must also keep records of the corrective actions that are not required to be reported to the FDA. Voluntary submission of such a report can be done based on 21 CFR 7
FDA’s Inspection Operation
On various grounds, the FDA performs inspections such as scheduled investigations, surveys, or a response to a reported problem. FDA’s Office of Regulatory Affairs (ORA) leads all such inspectional operations.
There are FDA Forms involved in these inspections,
Form 482 “Notice of Inspection” presented by the FDA immediately prior to inspection
Form 483 is issued to the firm’s management at the conclusion of the inspection if the investigator determines observations that violate the FD&C act rules
Objectives of Inspection after implementation of a Corrective Action
Check if the corrective actions implemented have been well defined and documented
Verify if the right sources of product and quality issues have been found
Check if there are any unfavourable trends in the product and quality information
Verification of the received data based on completeness, accuracy and time of reception
Check if appropriate statistical methods are applied to detect and identify the extent of recurring quality problems
Verify if a failure investigation is carried out and check if there has been a genuine attempt to find the root cause (wherever possible)
Check if there is control for the prevention of distribution of non-conforming products
Verify if appropriate actions have been taken for significant product and quality problems identified from data sources
The effectiveness of the corrective action is validified and verified before implementation by ensuring that the corrective actions do not lead to any adverse effects on the finished device
Check if corrective actions for product and quality problems were implemented and documented
Verify if all non-conforming product and quality problems and corrective actions have been properly disseminated, including dissemination for management review
FAQ
What is the Voluntary Malfunction Summary Reporting Program?
It permits manufacturers to report certain device malfunction medical device reports (MDRs) in summary form on a quarterly basis.
The VMSR program lets the manufacturers submit separate summary reports for each unique combination of the device model, brand name, device model, and problem code(s).
Every summary report is made available to the public in the MAUDE. It is mandatory to submit individual reports of death or serious injury events continue to be required, under sections 803.50 and 803.52, or 803.53, as applicable.
How to report Medical Devices licenced as biological products?
CBER (Centre for Biologics Evaluation and Research) is designated the lead centre in the FDA for regulating in vitro diagnostic (IVD) medical devices intended for screening or confirmatory clinical laboratory testing associated with blood banking practices and other process testing procedures.
IVD devices licensed as biological products are also subject to the applicable regulations under 21 CFR Part 803 – Medical Device Reporting.
Where to search for the medical device Reports?
The Manufacturer and User Facility Device Experience database contains mandatory reports filed by manufacturers and importers from August 1996 to the present, all mandatory user facility reports from 1991 to the present, and voluntary reports filed after June 1993.
The MAUDE database houses MDRs submitted to the FDA by mandatory reporters (manufacturers, importers and device user facilities) and voluntary reporters such as health care professionals, patients and consumers.
What are the data that are not required to be presented for routine review?
In accordance with Agency policy (CPG 7151.02), records regarding the results of internal quality audits, management reviews, third-party audits (including ISO audits), or supplier audits are not required to be presented.