Any action performed to reduce the risk of death or serious deterioration in health connected with the use of a medical device is referred to as Field Safety Corrective Action (FSCA). The manufacturer is required to take action to remove or limit the risk of the recognised dangers.
If a medical device malfunctions in Switzerland, the manufacturer is required to undertake an FSCA and Swissmedic keeps track of all FSCAs regarding medical equipment sold in Switzerland.
Reporting a Field Safety Corrective Action (FSCA)
Field safety corrective actions involving items placed on the Swiss market must be reported to Swissmedic by manufacturers
The Swiss authorised representative is responsible for reporting for manufacturers who are not headquartered in Switzerland
When a field safety corrective action is recorded, Swissmedic determines whether the risk can be effectively mitigated by the manufacturer’s steps and supervises their execution
The content is the responsibility of the manufacturer or an authorised representative (accuracy, completeness, and data protection)
Importers must immediately notify the manufacturer and its Swiss authorised agent of any complaints or reports of suspected incidents involving a device they have placed on the market
Distributors who receive complaints or reports about a device they sold must immediately notify the manufacturer, as well as the manufacturer’s Swiss authorised agent and the importer, if relevant
Healthcare professionals also must report serious occurrences to the supplier and Swissmedic
Such reports should be sent as soon as possible to the supplier organization with the help of the form available on the Swissmedic website.
A combined Initial-Final Report (combined initial-final MIR)
Evaluation by Swissmedic
Electronic submission
FSCAs must be reported with the help of the form released by Swissmedic. The Swissmedic website has this form available for download. This form must be used to send all FSCA reports to Swissmedic in an electronic, machine-readable manner.
Reports can be submitted in English or one of the official Swiss languages. All required fields must be filled out. The completed report form, the field safety notice (FSN), the customer list, and any other supporting paperwork should be emailed to [email protected].
If Swissmedic has any additional questions about an FSCA, it will contact you by email.
Timeline for reporting
If the serious incident clearly poses or has the potential to pose, a serious and imminent threat to the lives or health of a large number of people (serious public health threat), the report must be submitted as soon as possible, and no later than 2 calendar days after becoming aware of the incident
If the significant incident resulted in death or an unexpected serious worsening in a person’s health, the report must be submitted immediately, and at the latest within 10 calendar days
All other major incidents must be reported as soon as possible and no later than 15 calendar days after the date of discovery
An Initial Report must be made to Swissmedic if the manufacturer or Swiss authorised representative has not received sufficient information within the statutory time limit to determine whether or not a reportable occurrence has occurred
Types of Reports
1. Reporting of Trends
If a manufacturer sees a statistically significant increase in the frequency or severity of non-serious occurrences or expected negative side effects, they must notify SwissMedic by sending the Trends report form to mailto:[email protected].
The Swiss authorised representative is in charge of this role for producers who are not headquartered in Switzerland. The manufacturer or the Swiss approved representative might submit the report in this scenario.
2. Periodic Summary Report (PSR)
Serious incidents where the root cause is known (and/or) the serious incident is already the subject of an FSCA (and/or) the serious incidents occur frequently and are well documented then these incidents can be grouped and reported together to SwissMedic in the form of Periodic summary Report.
This process can be initiated by sending SwissMedic an email of this Periodic summary report form in English or any of the Swiss national languages.
3. Periodic Safety Update Report (PSUR)
Manufacturers of class IIa, IIb, and III medical devices must develop and submit a Periodic Safety Update Report (PSUR) for each product and, if relevant, for each product category or group, to the responsible notified body.
The PSUR and the Notified Body’s evaluation outcome must be submitted to Swissmedic upon request by the manufacturer or its Swiss authorised agent.
FAQ:
What is Field Safety Notice?
Every manufacturer is required to inform its users and, if applicable, patients about field safety remedial actions that have been implemented. Typically, this involves sending a Field Safety Notice (FSN).
The European Commission website has FSN templates as well as a confirmation form. The templates are designed to help manufacturers create high-quality customer letters that include all relevant information.
In the event of publishing, the FSN must not contain any information that would be in violation of data protection laws. Before publication, particularly sensitive personal data should be removed or, if absolutely necessary, anonymized.
Do Hospitals have to maintain a medical device vigilance system?
Yes, Hospitals are legally required to establish a reporting system. They must designate someone to be responsible for serious occurrences involving medical devices and they must publicly notify Swissmedic of this vigilant contact person for medical devices.
If there are any new vigilance contact persons for medical devices, or if the contact details of existing registered contact persons change, Swissmedic must be notified. Swissmedic inspects the reporting systems in hospitals.
Summary of Safety and Clinical Performance (SSCP) acts as a vital document that allows the public to access information quickly. The information in the SSCP can be sourced entirely from the technical file.
The technical file consists of the Post Market Surveillance (PMS), risk assessment, post-market clinical follow-up (PMCF) plans and reports. The SSCP document is required for high-risk devices only-this includes Class III and all implantable devices.
Manufacturers of custom-made or investigational devices need not produce this document. Implant card together with SSCP enables an efficient system to access device information.
SSCP for medical devices under MDR
Under MDD, the information on medical devices was not easily attainable. Therefore, the end-users of most medical devices were deprived of information regarding even high-risk medical devices.
As a result, medical devices under the directives lacked clarity. The main reason why SSCP is introduced is that MDR, unlike the directives, brings accessibility of information into account.
The SSCP should be made available on the EUDAMED website for easy access. It should be assigned an identifier that remains the same throughout the document’s lifetime. This identifier is not subjected to changes even when the content of this document is revised.
Language and readability requirements
SSCP follows the MDR language requirements like the other technical documents, such as Instructions for Use. All intended users within the EU understand no single language.
Therefore, language translations should be made available to intended users and patients. The SSCP should be translated into the languages accepted in the Member States where the device is intended to be sold.
Healthcare professionals widely understand English. Having an English translation available is essential, even if it is not available in selecting the official languages of each Member State. This enables the access to information that EU MDR strives to achieve.
While preparing SSCP, readability is an essential factor. The manufacturers must bear in mind to produce two sections. One part for intended users/healthcare professionals, and a second part for patients if applicable.
It is recommended to use an appropriate method to confirm that the document is understandable to the member of both categories. Further guidance on the readability, translations and other factors involved in SSCP can be found in the MDCG guidance on Summary of safety and clinical performance.
Sections of Summary of Safety and Clinical Performance
Article 32 of the EU MDR states some sections that are a must. The manufacturer may add further information from the Technical Dossier/File if relevant to the users.
The following sections are mandatory in an SSCP for healthcare professionals:
Details of the device and manufacturer (including UDI details).
Intended use of the device.
Description of the device and its components. Previous variants of the device.
Indications, contraindications, and target demographic.
Details of residual risks, undesirable effects, warnings, and precautions
Risks, undesirable effects, and warnings should be mentioned in SSCP. Other relevant aspects of safety, serious events, and a summary of any field safety corrective action must be included if applicable.
A summary of the Clinical Evaluation of the device.
This section is intended to summarise the clinical evaluation results and the clinical data, the evaluation of undesirable side-effects, and the benefit-risk ratio’s acceptability.
It is an objective and balanced summary of the clinical evaluation results of all the available clinical data related to the device. It should comprise favourable, unfavourable, and inconclusive data.
Conclusions based on evidence and the safety and performance of the device.
Suggested profile and training for users
Applied harmonised standards
All commonly applied specifications and international standards harmonised and adopted monographs should be listed in SSCP.
Revision history
Revision history should contain details such as revision validated by Notified Body (NB) and the language of SSCP validated.
A patient-specific SSCP template follows the same high-level structure as the clinician SSCP but does not include the reference to harmonized standards and common specifications. This decision focused on the information most relevant to patient health.
Validation of SSCP
When the Notified Bodies (NB) have assessed that all the required elements are included in the draft SSCP with the most current version of relevant documents in the TD, the SSCP has been validated by the NB.
SSCP validation may depend on the class of device and the conformity assessment routes chosen. More guidance on validation by NB can be found in the MDCG guidance on SSCP.
Uploading SSCP to EUDAMED
SSCP should be made available online for the users who intend to read the document. It is uploaded in EUDAMED by the Notified Bodies, the only actor managing the SSCPs in EUDAMED.
After each validation process, NB shall upload the updated SSCP by replacing the older version. However, once the ‘master’ SSCP is uploaded, it is up to the manufacturer to upload the translations to EUDAMED.
FAQs
What resources can be used for the SSCP?
The technical files like the design validation report, risk management report, clinical evaluation report (CER), as well as Post-Market Surveillance (PMS) and Post-Market Clinical Follow-up (PMCF) reports.
The Instructions for Use of the device can also be used as information for preparing the SSCP. Please note that SSCP cannot replace the IFU.
How frequently should SSCP be reviewed or updated?
The SSCP should be ideally updated annually. Documents like the PMCF evaluation and periodic safety update reports (PSUR) are updated annually. It is recommended to update SSCP along with the other technical documents.
How can I access the SSCP?
SSCP will be made available with the launch of the EUDAMED database. SSCP is accessible to both healthcare professionals and patients. SSCP has clear information for healthcare professionals with prior knowledge of medical terminologies.
In addition to this, SSCP should also contain a section that patients of different levels of expertise easily understand.
When should a manufacturer of Class III medical devices prepare SSCP?
SSCP should be made available at the time of registration.
Is there a similar requirement under IVDR?
Article 29 of In Vitro Diagnostics Regulation 2017/746 (EU IVDR) describes a requirement like the SSCP: the Summary of Safety and Performance (SSP).
The SSP requirements are almost identical to those of the SSP, replacing ‘clinical’ for ‘performance’ evaluation. Also, it includes a requirement for metrological traceability of assigned values intended for analytes used in IVDs.
For more information on IVDR, read our article on IVDR 2017/746.
On 5 May 2017, the EU published the new EU MDR 2017/745 and IVDR 2017/746 regulations in which they formally introduced the UDI system in the EU. The UDI comprises the following components
A device identifier (UDI-DI)
A production identifier (UDI-PI)
The Basic UDI-DI is a technique introduced by the EU for linking medical devices to their regulatory documentation so that the model of the product can be uniquely identified throughout its entire lifecycle.
The linked documentation may include the declaration of conformity, notified body certificates, technical documentation and a summary of the safety and clinical performance of the device.
The UDI-DI identifies specific, detailed information about a particular device. The UDI-DI is a unique numeric or alphanumeric code specific to a device model and used as the ‘access key’ to the information stored in a UDI database.
If any of the below details change, the device will then need a new UDI-DI.
Name or trade name of the device
Device version or model
Labelled as a single-use device
Packaged as sterile
Maximum number of uses
Need for sterilization before use
Quantity of devices provided in a package
Critical warnings or contra-indication
CMR/endocrine disruptors
Before placing a device for clinical or performance evaluation on the market, the manufacturer has to assign a Basic UDI-DI to the device and enter it into the UDI and Device Registration module of EUDAMED, along with other data elements related to that device.
The Basic UDI -DI should be maintained by the manufacturers by establishing processes for the assignment and maintenance of Basic UDI-DIs.
According to the new rules, any manufacturer can assign a UDI to the device and to all the higher levels of packaging of the device as well before the device is placed on the market. The label of the device should have the UDI carrier.
The manufacturer has to ensure that before the device is placed on the market, the information related to the device has to be submitted correctly and saved to the UDI database. In the EU, the manufacturer has to assign a UDI along with a Basic UDI-DI to all his devices.
Some important points on the UDI:
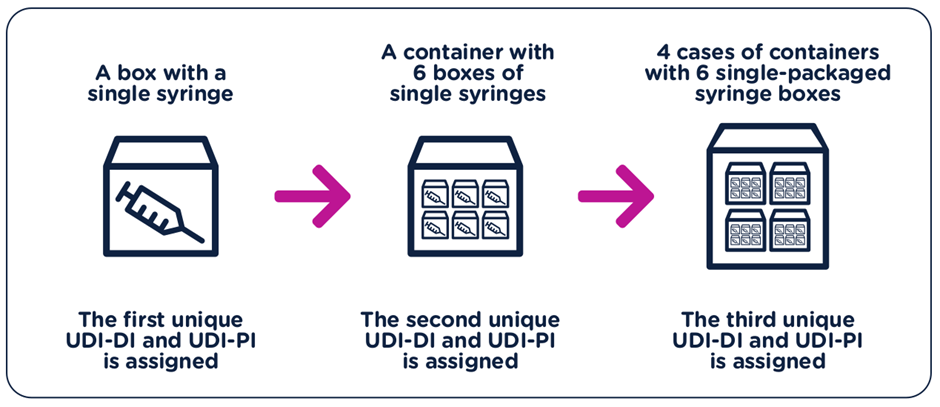
The UDI-DI have to be unique at each level of device packaging.
Each component considered to be a device and is also commercially available on its own must be assigned a separate UDI unless the components are part of a configurable device that is marked with its own UDI.
A new UDI-DI is required whenever there is a change that could lead to misidentification of the device or poses an error for its traceability.
The data for new UDI-DIs shall be available at the time the device is placed on the market.
Manufacturers shall update the relevant UDI database record within 30 days of a change being made to an element, which does not require a new UDI-DI.
Article 27 of 2017/745 and Article 24 of Regulation 746/2017 lay down that the UDI system shall consist
Of production of a UDI that comprises a UDI device identifier (‘UDI-DI’) specific to a manufacturer and a device, providing access to the information and a UDI production identifier (‘UDI-PI’) that identifies the unit of device production and, if applicable the packaged devices
Placing the UDI on the label of the device or its packaging
Storage of the UDI by economic operators, health institutions and healthcare professionals, in accordance with the conditions laid down in the article.
FAQ
What is the UDI?
The UDI is a series of numeric or alphanumeric characters created through a globally accepted device identification and coding standard.
It allows the unambiguous identification of a specific medical device on the market. The UDI is comprised of the UDI-DI and UDI-PI. The unique identifier may include information on the lot or serial number and be able to be applied anywhere in the world.
The production of a UDI comprises the following:
A UDI device identifier (‘UDI-DI’) specific to a device, providing access to the information laid down in Part B of Annex VI.
A UDI production identifier (‘UDI-PI’) identifies the unit of device production and, if applicable, the packaged devices, as specified in Part C of Annex VI.
What is the Basic UDI-DI?
The Basic UDI-DI is the primary access key for device-related information in the Eudamed database, and it is referenced in relevant documentation [e.g. certificates (including certificates of free sale), EU declarations of conformity, technical documentation, and summaries of safety and (clinical) performance]).
Its goal is to find and connect devices that have the same intended purpose, risk class, and key design and manufacturing features.
It is distinct from the device’s packaging/labelling and does not appear on any commercial item.
Any Basic UDI-DI must uniquely identify the devices (group) covered by the Basic UDI-DI.
What are the entities that provide the service of issuing UDI to medical devices?
GS1
Health Industry Business Communications Council (HIBCC)
ICCBBA
Informationsstelle für Arzneispezialitäten — IFA GmbH
How to get a Basic UDI-DI?
Basic UDI-DI should be assigned in compliance with the rules of the chosen issuing entity.
Can one Basic UDI-DI be linked with more than one UDI-DI?
Medical devices with the same intended purpose, risk class, essential design etc., are grouped under a single and unique Basic UDI-DI. As such, a medical device that has been assigned a UDI-DI can be linked to only one Basic UDI-DI. There is, therefore, a one-to-one relationship of 1 UDI-DI per 1 Basic UDI-DI. On the other hand, one Basic UDI-DI can be linked to multiple UDI-DIs. Therefore, there is a relationship of 1 Basic UDI-DI to N UDI-DIs.
Which products are subject to the UDI system?
The UDI system should apply to all devices, except custom-made and performance study/investigational devices.
Who is responsible for placing the UDI carrier on the device itself, on the label and the device’s package?
All UDI-related requirements are the responsibility of the manufacturer. This comprises assigning the UDI (and Basic UDI-DI), registering the UDI (and Basic UDI-DI) in the Eudamed database, and placing the UDI carrier on the device’s label, packaging, or, in the case of reusable devices, on the device itself.
What are the obligations of economic operators and health institutions in relation to UDI?
According to the two medical devices Regulations, manufacturers shall be responsible for the UDI assignment and placement of the UDI carrier, the initial submission and updates of the identifying information and other device data elements in the Eudamed database.
Manufacturers shall update the relevant database record within 30 days of a change being made to an element which does not require a new UDI-DI.
Distributors and importers shall verify that, where applicable, a UDI has been assigned by the manufacturer. All economic operators and health institutions shall store and keep preferably by electronic means the UDI of the devices, which they have supplied or with which they have been supplied if those devices belong to class III implantable devices.
Please note that the Commission may decide to adopt implementing acts to expand the scope of devices for which economic operators shall store and keep the UDI.
What is the procedure for systems and procedure packs to undergo a UDI registration?
Systems and procedure packs shall undergo a UDI registration, as described in MDR Article 29(2).
Before placing on the market a system or procedure pack pursuant to Article 22(1) and (3), that is not a custom-made device, the system or procedure pack producer shall assign to the system or procedure pack in compliance with the rules of the issuing entity, a Basic UDI-DI and shall provide it to the Eudamed database together with the other relevant core data elements listed in the MDCG 2018-4 guidance document.
Are devices, which are compliant with the Medical Device Directives (MDD and AIMDD) and placed on the market after the application date of the Regulations (legacy devices), continue to be subject to UDI requirements?
In order to facilitate the transition to the new system, the new regulations allow manufacturers to place products on the market after the general application dates of the new Regulations (and until 26 May 2024 at the latest) under valid Directive certificates.
These legacy devices are not subject to UDI obligations, but they should be registered in the Eudamed database.
Timelines for registration as described under question 6 also apply to these products. More information on the operational aspects of the registration of legacy devices is available in the MDCG 2019-5 guidance document.
How should a UDI appear on the label or package of a device?
The UDI Carrier [Automated Identification for Data Capture (AIDC) and human-readable interpretation (HRI) representation of the UDI] shall be on the label or on the device itself and on all higher levels of device packaging.
In the event of significant space constraints on the unit of use packaging, the UDI carrier may be placed on the next higher packaging level. Higher levels of packaging shall have their own unique UDI. Please note that shipping containers shall be exempted from the requirement.
The UDI must appear in a plain-text version/human-readable information (HRI) and in a form that uses AIDC technology. AIDC means any technology that conveys the unique device identifier or the device identifier of a device in a form that can be entered into an electronic patient record or another computer system via an automated process. The HRI consists of legible characters that people can easily read. If significant constraints limit the use of both AIDC and HRI on the label, only the AIDC format shall be required to appear on the label.
For devices intended to be used outside healthcare facilities, such as devices for home care, the HRI shall appear on the label even if this results in no space for the AIDC. For other specific requirements related to the UDI carrier, please consult Section 4 of Annex VI Part C of the two Regulations.
For single-use devices of classes I and IIa medical devices and class A and class B IVD medical devices packaged and labelled individually, the UDI carrier shall not be required to appear on the packaging, but it shall appear on a higher level of packaging, e.g. a carton containing several (individually packaged) devices. However, when the healthcare provider is not expected to have access, in cases such as in-home healthcare settings, to the higher level of device packaging, the UDI shall be placed on the packaging (of the individual device).
For devices exclusively intended for retail point of sale, the UDIPIs in AIDC shall not be required to appear on the point of sale packaging. If the UDI carrier is readily readable or, in the case of AIDC, scannable, through the device’s packaging, the placing of the UDI carrier on the packaging shall not be required.
What changes in the medical device would require a new UDI-DI?
A new UDI-DI shall be required whenever a change could lead to misidentification of the device and ambiguity in its traceability. In particular, a new UDI-DI shall be necessary in the case of any change of the following elements: name or trade name, device version or model, labelled as single-use, packaged sterile, need for sterilisation before use, the number of devices provided in a package, critical warnings or contra-indications and CMR/Endocrine disruptors. A UDI-DI shall be associated with one and only one Basic UDI-DI.
Is the software subject to UDI rules?
The UDI shall be assigned at the system level of the software. The only commercially available software and software that constitutes a device in itself shall be subject to that requirement. The software identification shall be considered the manufacturing control mechanism and shall be displayed in the UDI-PI. UDI requirements for software are laid down in Annex VI Part C of the two medical device Regulations. A dedicated guideline with additional information on this aspect is available in MDCG 2018-5 guidance document.
What is the mandatory deadline for a device to comply with the UDI requirements?
The obligation for UDI assignment applies from the date of application of the two new Regulations, i.e. 26 May 2021 for medical devices and 26 May 2022 for In Vitro diagnostic medical devices.
The obligation for submission of UDI data in the EUDAMED database applies from 26 November 2022 for medical devices and 26 November 2023 for in vitro diagnostic medical devices (provided that EUDAMED is fully functional before the date of application of the respective Regulation; otherwise, this obligation applies 24 months after EUDAMED has become fully functional)
However, manufacturers will be able to voluntarily comply with registration obligations from 26 May 2021 for medical devices and 26 May 2022 for In Vitro diagnostic medical devices. It shall be noted that, provided that Eudamed is fully functional, at any time after 26 May 2021 for medical devices and 26 May 2022 for In Vitro diagnostic medical devices, the complete registration of devices (Article 29 of MDR and Article 26 of IVDR) remains a pre-condition for the possible registration of their relevant serious incident in Eudamed.
The obligation for placing the UDI carrier applies according to the following timelines:
The device as per Regulation (EU) 2017/745 (MDR)
Implantable devices and Class III devices
Class IIa and Class IIb devices
Class I devices
Placing UDI-carriers on the labels of devices MDR Article 123(3)(f), Article 27(4)
26 May 2021
26 May 2023
26 May 2025
Direct marking of the reusable devices MDR Article 123(3)(g), Article 27(4)
26 May 2023
26 May 2025
26 May 2027
The device as per Regulation (EU) 2017/746 (IVDR)
Class D IVDs
Class C and B IVDs
Class A IVDs
Placing UDI-carriers on the labels of devices IVDR Article 113(3)(e), Article 24(4)
For implantable products, the manufacturer must provide the product information required under Article 16 of MedDO, the information required under Article 18 paragraph 1 EU-MDR and must include the implant card.
Healthcare institutions must enter the details of the implant recipient in the implant card and give the same to the recipient. They provide the essential information needed by the recipient in a quickly accessible form.
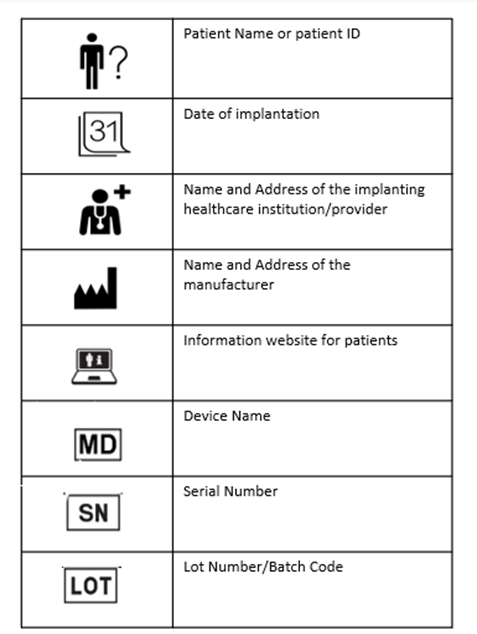
The implant cards will have the following details on them:
Device name
Device type
UDI or unique device identification
Serial /lot or batch number
Name and address of the manufacturer
Website of the manufacturer
Name of the patient or patient ID
Date of implantation
Name and address of the healthcare institution that performed the implantation
Purpose of an Implant Card
Implant cards aims to achieve three main objectives:
Enable the patient to identify the medical devices and to get access to vital information regarding the implant
Enable patients to identify themselves as persons requiring special care in relevant situations like security checks
Enabling emergency clinical staff or first responders to be informed about special care/needs for relevant patients in case of emergencies
The basic shape of the IC is designed in compliance with ISO/IEC 7810 ID-1 (credit card form). The symbols used already exist in the ISO standards and are hence widely accepted. The following picture provides an example for individual device ICs.
Not all implants need an IC. Some implants are exempted from the obligations laid down in Article 18 of EU MDR 2017/745. These include sutures, staples, dental fillings, dental braces, tooth crowns, screws, wedges, plates, wires, pins, clips, and connectors.
The information about this is in EU MDR Article 18 paragraph 3.
The UDI is a series of numeric or alphanumeric characters that is created through a globally accepted device identification and coding standard. It allows the unambiguous identification of a specific device on the market.
The UDI is comprised of the device identifier (UDI-DI) and the production identifier (UDI-PI). Both the UDI-DI and UDI-PI must be on the implant card.
The UDI Carrier (the representation of the UDI) is to be included on the IC in automatic identification and data capture (AIDC) format, e.g., linear or 2D-Barcodes, and the IC must include the UDI-DI in human-readable format. Source: Factsheet for manufacturers of implantable medical devices .