All medical devices are associated with inherent risks of some level. It is imperative to understand the medical device’s specific risks to a patient. Under EU MDR 2017/745, risk management is a continuous and iterative process.
Manufacturers are expected to plan, document, and implement risk management strategies in this process. These strategies may either eliminate the risk or mitigate the overall severity of the risk.
Medical Device Risk- Definition
As per Article II of EU MDR 2017/745, medical device risk is defined as ‘the combination of the probability of occurrence of harm and the severity of that harm’.
According to the definition, the strategies help prevent particular harm or risk and prevent severe harm.
Risk Management under MDR
Annex I section 3 of EU MDR 2017/745 mentions its requirements specific to the European medical device regulations. Manufacturers, under MDR, must implement the following aspects of risk management to be fully compliant.
Establish and document its plan for each device
Identify the known and foreseeable hazards associated with the device
Estimate and evaluate the risks associated with, and occurring during, the intended use and during reasonably foreseeable misuse
Eliminate or control the risks
Evaluate the impact of information from the production phase to the post-market phase on hazards and the frequency of occurrence of associated risks, the overall risk, benefit-risk ratio, and risk acceptability
Amend risk control measures if necessary
While implementing risk control measures to design and manufacture devices, the following aspects must be considered. Manufacturers must:
Eliminate risks through safe design and manufacture of the device
Take adequate protection measures (such as including alarms) if the risks cannot be eliminated
Provide information for safety (warnings/precautions/contra-indications) and training to users.
Certain medical device risks may be due to device usage errors. In Annex I Chapter I, MDR clearly states that such risks can be prevented by:
Reducing risks related to the ergonomic features of the device and the environment in which it is intended for use
Consideration of technical knowledge, experience, education, training and use environment, and the medical and physical conditions of intended users
How are device risks managed?
Risk management can be considered a 5-step procedure.
Step 1: Risk management plan
All these activities must be planned. The plan lays forth a strategy for risk management activities to be carried out throughout the product lifecycle.
This plan is documented in a risk management file containing the risk management plan and a risk management report.
Step 2: Risk assessments
Risk assessments evaluate the risk identified in normal and abnormal medical device use. Normal use of a medical device is the intended application of the device following all instructions by the manufacturer.
In contrast, abnormal use is when the medical device was used, violating the device instructions.
Step 3: Risk Control
Risks are controlled by implementing its plan. The risk-control measures chosen must be executed, and their effectiveness must be validated. This is done for an effective quality management system.
Step 4: Evaluation of residual risks
Complete elimination of risk may not be possible all the time. Therefore, it is imperative to identify the residual risk so that small and expected rather than massive, unexpected risks.
Step 5: Risk management review
As risk management is an iterative process, reviewing the risk control measures adopted and their effectiveness is imperative. This is ensured by post-market surveillance systems, clinical evaluation, and vigilance systems.
Maintaining updated risk systems and documents constitutes an effective quality management system for any medical device.
FAQs
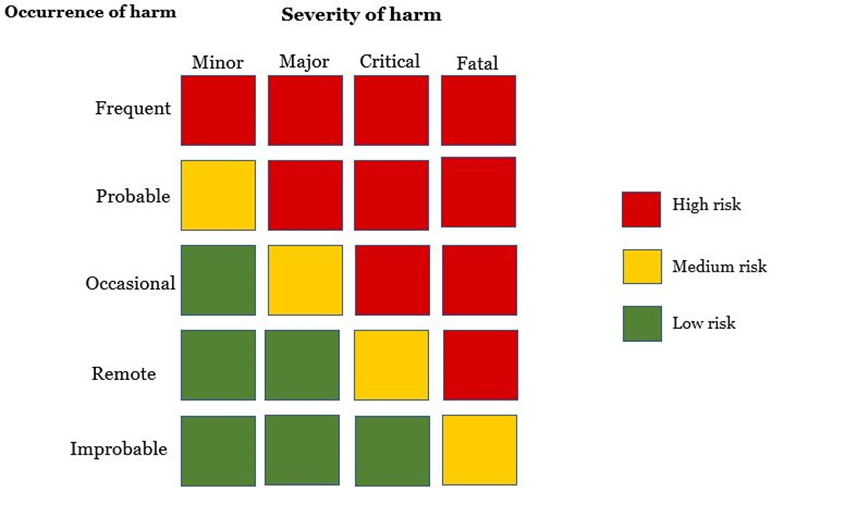
How are risks categorised?
Risks are classified based on the occurrence and severity of harm caused. The figure below is a risk matrix used to illustrate a matrix on all foreseeable risks. This is useful for evaluating residual risks posed by the medical device on the patient.
What is the EU MDR harmonized standard adopted for Risk Management?
EU MDR has adopted ISO 14971 for the Application of risk management to medical devices. This ISO standard allows manufacturers to identify hazards of a medical device and implement control measures for the same.
What is the role of Risk management in a clinical evaluation procedure?
Clinical evaluation is imperative to risk management as this allows the manufacturer to identify all possible risks associated with the device. This data can be used for the identification of safety concerns and appropriate risk management methods can be implanted. In other words, clinical evaluation is one of the inputs to risk management.
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.
The revolution in the digital sector has resulted in the Internet of Things (IoT), Software as a Medical Device (SaMD), Internet of Medical Things (IoMT) and other connected devices permeating the healthcare environment, both in hospital and home, has ended up with the possibility of cyberattacks and intrusions against the connected medical devices and the networks to which such a device is connected.
Most Medical devices are connected to the Internet, hospital networks, and other medical devices to provide health care and increase the ability of healthcare providers to treat patients.
These features also increase potential risks for Cybersecurity. Medical devices, like other computer systems, are vulnerable to security breaches, potentially impacting the safety and effectiveness of the device.
Since 2005, the FDA has tried to accomplish and enhance medical device cybersecurity, and the latest FDA effort is to create draft guidance that expects security throughout the total product life cycle (TPLC).
Another effort is the Protecting and Transforming Cyber Health Care Act of 2022 (PATCH Act of 2022),which, if passed, would revise the existing Federal Food, Drug, and Cosmetic Act.
The FDA guidance establishes six broad expectations on the Secure Product Development Framework (SPDF), which covers all aspects of a product’s life cycle, for the development, release, support, and decommission and satisfy Quality System Regulations (QSR) under 21 CFR Part 820:
Cybersecurity is a fundamental part of device safety and the QSR
Security by design
Transparency
Security risk management
Security architecture
Testing/objective evidence
The FDA draft guidance, under QSR, also declares that verification and validation activities by the medical device manufacturers shall include sufficient testing performed on the Cybersecurity of the system, which validates their inputs and outputs.
Further, the FDA summarizes that cybersecurity controls require testing beyond standard software verification and validation to demonstrate that the device has a good assurance of safety and effectiveness.
The following cybersecurity testing and corresponding objective evidence would be considered as the minimum support for a premarket submission:
Security requirements
Evidence of their boundary analysis creates a rationale for their boundary assumptions.
Threat mitigation
Evidence that all the design input security requirements were implemented successfully
Evidence of testing their threat models that demonstrates effective risk control measures provided in the system and use case
Evidence of the adequacy of risk control.
Vulnerability testing – Evidence on the testing of malformed
Abuse case and unexpected inputs
Vulnerability chaining
Closed box testing of known vulnerability scanning
Software composition analysis of binary executable files
Static and dynamic code analysis
Penetration testing– Identify and characterize security-related issues that discover security vulnerabilities in the product.
Regular interval cybersecurity testing – It is performed at regular intervals to identify the potential vulnerabilities before exploitation
This draft guidance replaces the 2018 draft version, which emphasizes the importance of understanding that all medical devices are designed securely, enabling new cybersecurity risks to be mitigated throughout the Total Product Life Cycle, and it elaborates the outline of the FDA’s recommendations more clearly for premarket submission to address cybersecurity concerns.
03/08/2022 Cybersecurity Alert: Vulnerabilities identified in medical device software components: PTC Axeda agent and Axeda Desktop Server
The PTC Axeda agent and Axeda Desktop Server are cloud-based technologies that allow people to securely view and operate the same desktop through the Internet. The Axeda agent and its desktop server are owned by the computer software company PTC.
The FDA alerts all medical device users and manufacturers about a cybersecurity vulnerability identified for the Axeda agent and Axeda Desktop Server.
The agent and desktop server of Axeda are used in many medical devices across several medical device manufacturers, and all the versions of the Axeda agent and Axeda Desktop Server are affected.
On the 8th of March, 2022, the Cybersecurity and Infrastructure Security Agency (CISA) published an advisory, ICSA-22-067-01, on these vulnerabilities.
Any successful exploitation of this vulnerability could allow an unauthorized attacker to take complete control of the host operating system, resulting in full system access, remote code execution, reading or changing the configuration, system file access, accessing log information, and other denial condition.
These vulnerabilities may result in changes to the functions of the medical device and impact the availability of the remote support functionality.
As a result, PTC recommends that affected manufacturers:
To upgrade Axeda agent Version 6.9.2 build 1049 or 6.9.3 build 1051 while running older versions of the Axeda agent.
Also, to configure the Axeda agent and Axeda Desktop Server to listen only on the local host interface 127.0.0.1.
Then, Provide a unique password in the AxedaDesktop.ini file for each and every unit.
Remove the installation file.
Make sure to delete the ERemoteServer file from the host device.
Never use ERemoteServer in production.
When running the Windows operating system, first configure Localhost communications (127.0.0.1) between ERemoteServer and Axeda Builder.
When running in Windows or Linux, only allow connections to ERemoteServer from trusted hosts and block all others.
Configure the Axeda agent for the authentication information required to log in to the Axeda Deployment Utility.
So, Cybersecurity is one of the crucial aspects of today’s fast pacing digital world. The threats caused by Cybersecurity, especially on medical devices, are hard to deny. It is important to learn how to defend themselves from them and create a safe environment for the usage of medical devices.
EU MDR and IVDR
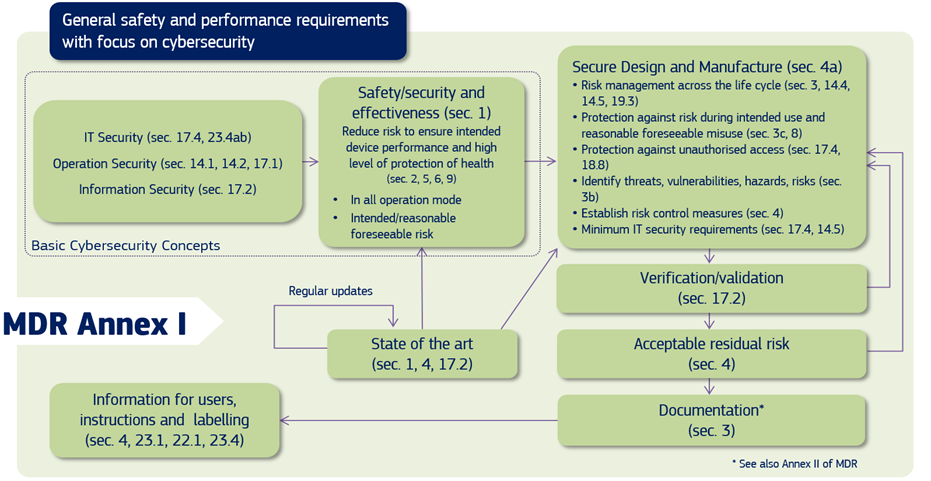
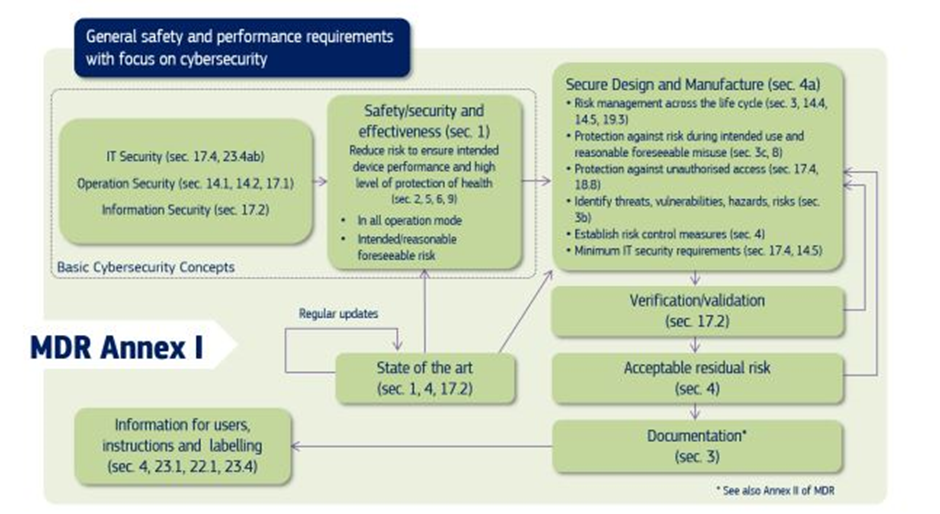
In the EU, both the MDR and IVDR Annex I create requirements for mandate consideration of medical device cybersecurity, and the Medical Device Coordination Group (MDCG), in its guidance, explains to the manufacturers of medical devices how to fulfil all the relevant essential requirements regarding Cybersecurity.
Source: MDCG 2019-16 Guidance on Cybersecurity of medical devices Figure 1: Cybersecurity requirements contained in MDR Annex I
The NIS Directive also provides for legal measures to increase the overall level of Cybersecurity in the EU.
GDPR (General Data Protection Regulation) helps the manufacturers of medical devices in regulating, protecting and processing personal data by the individual, company or organization that relates to the EU.
The EU Cybersecurity Act certifies Cybersecurity for ICT products, services, and processes.
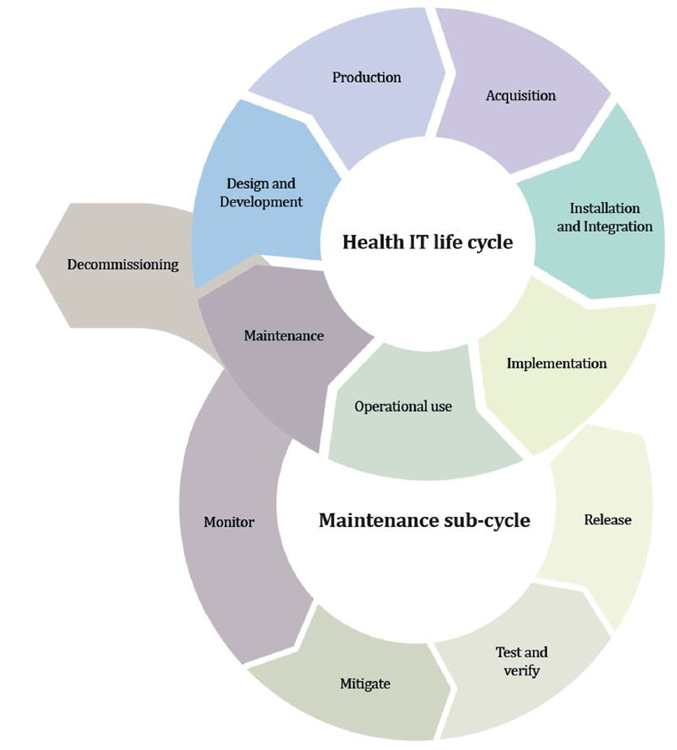
According to the Cybersecurity Act, manufacturers are required to demonstrate state of art in the design, development, and improvement of their medical devices throughout their life cycle.
During that period, the manufacturers must consider the safety, security, and efficacy of the medical devices, and in vitro diagnostic safety mechanism design must be considered early during the manufacturing process.
Source: MDCG 2019-16 Guidance on Cybersecurity of medical devices Figure 4: Lifecycle stages
The MDCG has proposed some key philosophies of the staged security concept strategy (“defense in depth strategy”) as follows:
Security management
Specification of security requirements
Security by design
Secure implementation
Management of security-related issues
Security update management
Security risk management
The list of possible IT security requirements for the operating environment according to MDCG:
Compliance with national and EU regulations (e.g., GDPR).
Ensuring appropriate security controls are in place
Ensuring the physical security of the medical device through security measures
Ensure control and security of network traffic through proper measures
Life Cycle Aspects
Security measures specific to their workstations connected to the medical device.
Security vulnerabilities related to the device hardware/software and third-party hardware/software used with the medical device.
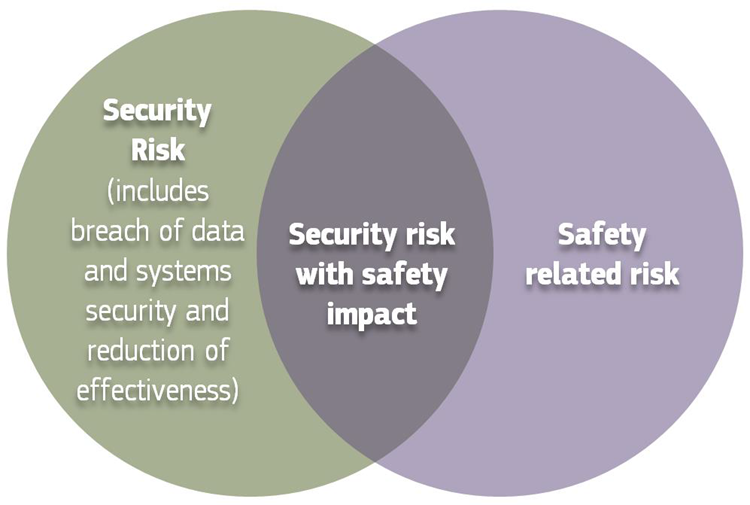
During the life of the devices, the manufacturer should implement the process to collect post-market information about the security of the device.
Source: MDCG 2019-16 Guidance on Cybersecurity of medical devices Figure 3: Cybersecurity measures may cause safety impacts
Based on the EU Cybersecurity Act, the manufacturer must provide the following information to the user of the medical device:
Specifications of the operating system
IT security risk assessment information.
Provisions for ensuring the integrity of software updates and security patches
Product installation
Security configuration options
Initial configuration guidelines
Step-by-step instructions for deploying security updates
Description of the backup and restore functions for data and configuration settings
Procedures for using all the medical devices in failsafe mode
The manufacturers are required to establish a post-market surveillance (PMS) system and actively keep these PMSs (Post Market Surveillance) up to date. Medical device cybersecurity requirements should be part of this PMS system.
Depending on the class of medical device, a PMS report or PSUR report will be generated, which concludes the analysis of all data from the market.
FAQs
How can we protect heath care from cyber-attacks?
· Vulnerability assessment and required testing · Training health care providers to protect from any breaches · Follow the standards of the regulations
Where is Cybersecurity used?
Cybersecurity helps in protecting the Datas, software or hardware connected with the system. This reduces unauthorized access to the data or the system.
What is the PATCH act?
PATCH act helps to meet all the Cybersecurity requirements for the manufacturer to complete FDA regulation standard.
What medical devices can be hacked?
MRI, Pacemakers, Implants, Heart rate monitors, Drug infusion pumps, medical records and other devices connected to the hospital network.
What are the new cybersecurity requirements according to EU MDR?
MDR Annex I explain the risks associated with the interaction between software and medical devices. Manufacturers should follow standard during life cycle, risk management, verification, and validation of the devices.
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.
EU Cybersecurity laws for Medical devices are advancing, and the use of software medical devices is also increasing daily. The increased interconnection of medical devices to computer networks and technological convergence have made devices and software programmes vulnerable to mishaps.
The importance of protecting patient data from cyber-attacks is now well recognised. With the advancement of software as a medical device, proper regulations must be established to ensure the safety and security of medical devices.
Read our article on SaMD regulations in the EU and UK to understand software medical devices. This article discusses the cybersecurity aspects of medical devices.
Why is cybersecurity important for medical devices?
Medical devices contain crucial patient information. Healthcare data has been the most common target for data breaches for over a decade. These data breaches contribute to the data leak; even patient life can be in danger due to outdated software.
EU Cybersecurity Laws for medical devices
Within the EU, the following legislative acts apply concurrently to the Medical Devices Regulations. These are important to the cybersecurity of medical devices or operators dealing with the protection or processing of personal data held in medical devices:
NIS Directive or Directive 2016/1148 of the European Parliament and of the Council of 6 July 2016 concerning measures for a high common level of security of network and information systems across the Union
GDPR (General Data Protection Regulation) or Regulation (EU) 2016/679 of the European Parliament and the Council on the protection of natural persons regarding the processing of personal data and the free movement of such data
EU Cybersecurity Regulation or Regulation (EU) 2019/881 of the European Parliament and the Council on ENISA (the European Union Agency for Cybersecurity) and information and communications technology cybersecurity certification and repealing Regulation (EU) No 526/2013 (Cybersecurity Act)
NIS Directive or Directive 2016/1148 aims to achieve cybersecurity in the EU by ensuring the following aspects:
Increase the preparedness of Member states by requiring them to be appropriately equipped
Setting up a cooperation group, there is cooperation among the Member States. This includes setting up of a Computer Security Incident Response Team (CSIRT) and a competent national NIS authority
A custom of security in all vital economic sectors like banking, energy, transport, etc
GDPR (General Data Protection Regulation) or Regulation (EU) 2016/679 governs the processing of personal data belonging to individuals in the EU. Personal data is any information used to identify or find a living person. Many parts of information that, when gathered, can lead to the identification of a specific person constitute personal information.
EU Cybersecurity Regulation or Regulation (EU) 2019/881 establishes European Cybersecurity Certification Framework for ICT products and services and specifies the tasks of the European Union Agency for Network and Information Security (ENISA) in the field of cybersecurity.
In addition to the above, it is imperative to follow the International Medical Device Regulators Forum IMDRF guidelines.
EU MDR Requirements on Cybersecurity
Specific cybersecurity requirements for medical devices are mentioned in Annex I of EU MDR 2017/745. The following flowchart summarises the cybersecurity requirements mentioned in Annex I.
The following MDR provisions list is applicable for all medical devices. The list applies to software medical devices as well. The documentation requirement is the same for medical and software medical devices, but the document’s content varies.
Conformity assessment procedures: Article 52
Post-market surveillance (PMS) system, PMS plan and report: Article 83-85
Periodic safety update report: Article 86
Reporting of serious incidents and field safety corrective actions: Article 87
Trend reporting: Article 88
Analysis of serious incidents and field safety corrective actions: Article 89
Technical documentation: Annex II and Technical documentation on post-market surveillance: Annex III
Clinical evaluation and post-market follow-up: MDR Chapter VI and Annex XIV
FAQs
Are labels required for software medical devices?
Yes, software medical devices are required to have appropriate labels. It is essential to convey to the end-user the relevant information. This is done by including labelled information on potential risks associated with the product, preventive measures to be taken and any other relevant information for the end user. As per the IMDRF guidance document, labels should include the following information: · Device instructions and product specifications for the intended use environment · Description of backup features · Guidance to users regarding supporting infrastructure requirements for the device to operate as intended. · A description of how the device is protected or can be protected using a secure configuration. Secure configurations may include anti-malware · Complete list of network ports and other interfaces of the device · Detailed system diagrams for end-users. · Where appropriate, risks of using the medical device outside of the intended use environment · A description of procedures for download and installation of updates Annex I Section 23.2 of EU MDR 2017/745specifies labelling requirements. Some of the EU MDR 2017/745 requirements include:
· Trade name or product name · Manufacturer name · Address of registered place of business · Precaution or warnings that require the immediate attention of end-user · Any other relevant information regarding the product
Do software medical devices require an Authorised Representative?
Software medical devices are not exempt from this requirement. An AR must be appointed if the manufacturer is based out of the European Union. All obligations of AR mentioned in Article 11 of the EU MDR 2017/745 apply.
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.
The SaMD definition statement should include a clear and robust statement about intended use, including the following:
The term “Software as a Medical Device” (SaMD) is defined as software intended to be used for one or more medical purposes that perform these purposes without being part of a hardware medical device.
SaMD is software that carries out one or more medical tasks. If it might be incorporated with a piece of hardware, the software performs a medical function.
The international medical device regulators Forum (IMDRF) defines SaMD as “software that may operate on general-purpose (non-medical) computing platforms, may be combined with other products, including medical devices, and may interface with other medical devices or other general-purpose hardware or software that provide input to SaMD.”
Software that turns the magnets in an MRI or animates an X-ray control panel are examples of software that is necessary for hardware operation but is exempt from SaMD. Software that only organizes data, retrieves data, or speeds up operations is also insufficient.
7 Classification of IMDRF Regulation on Software as a Medical Device
The following are necessary principles required in the categorization approach of SaMDs.
An accurate SaMD definition is a prerequisite for the categorization of the device.
The importance of the information provided by the SaMD to the healthcare decision and the situation or condition are combined to determine the categories.
The four classifications (I, II, III, and IV) are based on the severity of the impact on the patient or public health, where accurate information provided by the SaMD to treat or diagnose, guide or inform clinical management is essential to prevent death, long-term disability, or other severe deterioration of health, mitigating public health.
The impact level is at its maximum in Category IV and lowest in Category I.
According to the details provided in the SaMD definition statement, a manufacturer’s SaMD is classified in the highest category when it can be employed in various healthcare settings or conditions.
The classification of SaMD should be correctly re-evaluated when a manufacturer modifies SaMD during the lifecycle that causes the definition statement to change. The information in the revised (new) SaMD definition statement is used to categorize the SaMD.
Even when a SaMD is interfaced with other SaMDs, other hardware medical devices, or employed as a module in a more complex system, it falls under the category specified in its SaMD defining statement.
State of Healthcare situation or condition
Significance of information provided by SaMD to healthcare decision
Treat or diagnose
Drive clinical management
Inform clinical management
Critical
IV
III
II
Serious
III
II
I
Non-serious
II
I
I
The above table represents the risk categorization of IMDRF regulation for SaMD based on the significance of the information provided by SaMD to healthcare decisions and the state of the healthcare decision.
Identifying risks and putting safeguards in place that assure acceptable risks are necessary for creating safe SaMD. It is well acknowledged that testing software is insufficient to establish operational safety.
As a result, it is acknowledged that confidence should be included in the software to guarantee its security.
The IEC 62304 standard governs the lifecycle development of medical device software. The standard highlights three key safety concepts that are pertinent to SaMD, defines basic testing requirements, and proposes a risk-based decision methodology:
Risk management.
Quality management.
Methodical and systematic systems engineering according to best industry practices.
Combining these concepts helps IMDRF regulation on SaMD manufacturers follow a clearly structured and consistently repeatable decision-making process to promote safety for SaMD.
Design and Development
A quality-assured approach to software development should consider the selection and implementation of system design and development methods that:
Incorporate a structured development using models, methods, architecture and techniques appropriate for the development language(s) and the device’s intended use,
Cover the various stages of the software lifecycle using IEC 62304 as a reference standard for software development. Software Engineering Body of Knowledge (SWEBOK) and Systems Engineering Body of Knowledge (SEBoK) must be used as reference books for software engineering and
The design and development process must be documented systematically (using appropriate tools.)
Post Market Surveillance
SaMD manufacturers should continuously monitor customer complaints to maintain the safety level because software hazards can never be eradicated.
Customer feedback must be gathered as part of the monitoring process, for instance, through enquiries, grievances, market research, focus groups, service, etc. SaMD and other software have inherent characteristics that effectively comprehend and record user experiences.
The use these feedback mechanisms by IMDRF regulation on SaMD makers is advised to comprehend failure modes and conduct analysis to address safety concerns.
Additionally, it is recommended that SaMD manufacturers expand their monitoring to detect system or software errors automatically, i.e., identify and fix an error before a failure happens.
Quality Management System
Medical device QMS principles allow the measurement of activities depending on
The type of medical device.
Risk of the product to patients.
Size of the organization.
Technology or automation is used to manufacture.
And other factors are determined by the manufacturer to control quality and maintain the safe and effective performance of the medical device.
SaMD, a software-only product, is manufactured primarily using automated software development tools to assist development lifecycle activities.
In some circumstances, these computerized processes could take the role of deliberate or discrete acts generally present in creating physical items (such as transferring design to production).
To control the quality of SaMD, however, it is still necessary to adhere to the QMS principles that give the lifecycle processes and activities structure and support.
An effective QMS for IMDRF regulation on SaMD have to include the following principles:
An organizational structure ensures SaMD’s safety, efficacy, and performance by providing leadership, accountability, and governance with enough resources.
A set of SaMD lifecycle support processes that are scalable to the organization’s size and applied consistently across all realizations and use processes
A set of realizations and use processes that are scalable for the type of SaMD and the organization’s size takes into account essential elements required for assuring the safety, effectiveness, and performance of IMDRF regulation on SaMD.
FAQs
What are some of the examples of SaMD?
· Applications for viewing MRI, Ultrasound, or X-Ray examinations images · Software for detecting cancer using image processing · Software using AI for analysis of test reports and detection of pathologies · Software that analyses data from other devices for assessing the patient health and calculating the risks · Applications that calculate the dosage of the patient using the data of patient health
How do I know if my application is classified as SaMD?
Classification of software as a medical device can be tricky. However, it is imperative first to know if the software falls under the definition of SaMD. One should consider the following to classify any software as a medical device: · If the software can perform or take part in the treatment processes · It can be launched on hardware devices such as smartphones, computers, tablets, smartwatches · If the software is not intended to manage a hardware medical device and is a standalone medical product · It can be used with hardware medical devices and integrated into the ecosystem of devices If the above is invalid, the software does not fall under a medical device, and medical device regulations need not be followed.
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.
Technical documentation should contain details of the medical device in a clear, organized, readily searchable and unambiguous manner.
It should be provided with all medical devices irrespective of the device class and to be kept updated throughout the product life cycle. Technical documentation is to be prepared according to the requirements given in Annex II of EU MDR 2017/745.
Requirements of Technical Documentation as per Annex II
Device description and specification, including variants and accessories
Information to be supplied by the manufacturer
Design and Manufacturing information
General safety and performance requirements
Benefit-risk analysis and risk management
Product verification and validation
Device Description and Specification, including variants and accessories
This section mentions that technical documentation must include all basic device details such as trade name, general description, basic UDI-DI, principles of operations, classification details, accessories description, key functional elements, technical specification etc. There must also be a proper reference to previous or similar medical devices.
Information to be supplied by the Manufacturer
This section mentions that manufacturer must include a complete set of labelling for the device and its packaging. The manufacturer must also ensure the language requirements of the same according to the each member states’ language requirements.
Design and Manufacturing Information
This section mentions that the technical documentation shall include complete information on processes carried out by the manufacturer during the design stage, manufacturing, validation, monitoring and final product testing of the device.
Identification details of all sites and people involved in the manufacturing process must also be made available.
General Safety and Performance Requirements
This section mentions that the technical documentation must demonstrate conformity to general safety and performance requirements set out in Annex I of the MDR.
This shall include details such as an explanation for safety and performance requirements that the device satisfies and an explanation for requirements that it does not meet, methods used to demonstrate conformity, harmonized standards, Common specifications, and the identity information of the control documents.
Benefit-Risk Analysis and Risk Management
This section mentions that the technical documentation shall include the benefit-risk analysis, solutions adopted, and risk management results as per Annex I of the MDR.
Some significant points from the Risk management section of Annex I are
The risk management plan must be available for each device
Should be able to identify foreseeable hazards associated with each device
should be able to estimate and evaluate the risks
should have plans to eliminate/control the risks
should be able to assess the impact of risk at various stages of the device’s lifecycle and thereby estimating overall risk, benefit-risk ratio, and risk acceptability
Section 1 to 8 of Annex I gives detailed information on general safety measures and other risk management requirements.
Product Verification and Validation
This section mentions that the technical documentation shall contain results and critical analyses of all verification and validation tests/studies.
Some significant Tests/studies discussed in the section are
Biocompatibility
Electrical Safety
Electromagnetic compatibility
Software verification and validation
Stability
Performance and Safety
Physical, chemical, and microbiological characteristics
sterility
A clinical evaluation report and a PMCF (Post-market clinical follow-up) plan and evaluation report must be available. Else, a justification for why PMCF is not applicable must be made available.
Additional information required in specific cases is given in detail in Section 6.2 of Annex II of the EU MDR.
Post-Market Surveillance
Annex III of the MDR majorly mentions that the post-market surveillance plan must address the method of collection and utilization of available data (Such as information on serious incidents and their corrective actions, data on any undesirable side- effects and so on) and includes a list of details a post-market surveillance plan must include in the plan.
There is also a reference to PSUR (Periodic safety update report) and post-market surveillance report mentioned in articles 86 & 85, respectively.
Significant Changes to be noted during the transition from MDD to MDR
Device description to include UDI, UDI-DI or any such number for traceability reasons
Reference requirements to previous or similar medical devices
Under labelling requirements in the MDR, it is explicitly talks about the labelling of devices and their packaging
Language requirements for labelling
As per the MDD, only Class III devices required an explanation for design stages and procedures. The same is now updated to all classes of medical devices in MDR
Identification requirements of all manufacturing sites, design including suppliers and contractors are required now
General safety and performance requirements are updated versions of Essential requirements from the MDD
The benefit-risk analysis is explicitly mentioned as part of risk management requirements in the MDR
Product verification and validation requirements
A detailed explanation of pre-clinical and evaluation and Clinical Evaluation requirements are provided in the MDR
The requirements of the Clinical Evaluation plan and report, Post-market clinical follow-up plan and report are specially mentioned in the MDR
Additional information for special case devices (e.g., combination device, sterile device, device with measuring function etc.) is mentioned in detail in the MDR
FAQs:
What are Common specifications?
Common Specifications are a set of technical and/or clinical requirements, other than a standard, that provides a means of complying with the legal obligations applicable to a device, process or system.
What is the UDI?
The UDI is a series of numeric or alphanumeric characters that is created through a globally accepted device identification and coding standard. It allows the unambiguous identification of a specific medical device on the market. The UDI is comprised of the UDI-DI and UDI-PI. The unique identifier may include information on the lot or serial number and be able to be applied anywhere in the world.
What is the Basic UDI-DI?
Basic UDI-DI is intended to identify and connect devices with the same intended purpose, risk class and essential design and manufacturing characteristics. It is independent/separate from the packaging/labelling of the device and it does not appear on any trade item
Is Post-market surveillance (PMS) applicable for only higher risk classes?
PMS is required for all device classes. A PMS plan and report to be maintained and kept updated by the manufacturer throughout the device life cycle.
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.
Medical devices conforming with the medical device regulations must have a conformity marking. Medical devices that do not conform may still be placed in the market, provided they apply through the exceptional use devices pathway.
This article discusses the exceptional use requirements to be satisfied to gain access to EU and UK markets.
In the UK, the UKCA marking displays that the device conforms to UK Medical Device Regulations 2002. Without UKCA marking, the devices can only be placed in the market under certain circumstances.
Similarly, CE marking is accepted throughout the EU, which denotes that medical device conforms to the EU MDR 2017/745. This procedure may also apply to custom-made devices that have not complied with standard conformity procedures.
These devices are called special-purpose devices in the EU and are mentioned in Article 21 of EU MDR 2017/745.
The application form must be submitted for each different patient. MHRA approves the case-by-case application basis and may request more information if the application details are deemed unclear.
The application form for exceptional use devices requires the following details:
Risk analysis, hazard identification and estimation of risks
Name and address of the consultant
Patient’s medical condition details
Device necessity and
Other relevant information
Devices for special purposes in the EU
Devices under this category do not require a CE mark. Still, they must meet the conditions mentioned in EU MDR that the device is evaluated for its safety and effectiveness by the manufacturer.
Articles 62-82 of EU MDR describe the requirements to be met by manufacturers of investigational devices.
Firstly, for investigational devices, a proper clinical investigation plan is required. Read our detailed article on Clinical investigation in the EU for more information regarding this topic.
In short, the clinical investigation has a different technical file and application form than the CE-marked medical devices.
The application form should be submitted with the following documents.
Clinical investigation plan
Investigator’s brochure
Name and address of the manufacturer
Patient information sheet
Detailed risk analysis and test reports
Informed consent document
Other available technical documentation
Custom-made devices
As per EU MDR, ‘custom-made devices’ means as any device specifically made in accordance with a written prescription of any person authorised by national law by virtue of that person’s professional qualifications which gives, under that person’s responsibility, specific design characteristics, and is intended for the sole use of a particular patient exclusively to meet their conditions and needs.
However, the definition does not include ‘mass produced’ devices that need to be adjusted per user requirements. UK MDR 2002 5 (1) has a similar definition for custom-made medical devices.
The provisions in Great Britain (England, Wales and Scotland) can be accessed in Guidance on Custom-made devices in GB. Medical devices like dental appliances and prosthetics all come under custom-made devices as these are designed specifically for an individual and are not mass-produced.
Custom-made devices require technical documentation following Annex II and III of EU MDR. In addition to the documents, the manufacturer or authorised representative should draw a statement containing crucial information like that the device is intended for exclusive use by a particular user and data allowing unique identification of the medical device.
An important point to remember is that custom-made devices must conform to general safety and performance requirements in Annex I of EU MDR and essential requirements in Annex I Schedule 2A of the UK MDR 2002, and the devices are not exempt from these requirements.
FAQs
What devices come under custom-made devices?
Medical devices that come under Custom-made devices are specifically designed for an individual patient. Some great examples include dental crowns, fillings, elastics and retainers for dental structure or health. Other devices such as artificial eyes and maxillofacial implants like craniofacial implants, auricular, nasal implants, hearing aids and orthopaedic footwear fall into this category. The healthcare professionals must take accurate impressions of patients so that the device is a custom fit for the end-user or patient.
What is the difference between a custom-made device and an investigational device?
Investigational devices are those that are research devices that are developed that have no market equivalent. These devices are subjected to a clinical study to check product effectiveness. On the other hand, custom-made devices are not subject to clinical research but are customised according to the patient’s requirements. Both investigational devices and custom-made devices do not require CE/UKCA marking and require exceptional devices regulatory approvals to enter the respective markets.
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.