Facing a medical device audit can be daunting, but with meticulous preparation and strategic responses, companies can turn this challenge into an opportunity for building a robust quality system.
Tips for Mastering Medical Device Audits
This article provides a detailed roadmap for mastering medical device audits, covering essential steps from internal audits to adeptly handling regulatory findings.
1. Demystifying Audits
Understanding the fundamental concepts behind medical device audits is crucial. ISO 19011 defines audits as systematic, documented, and independent processes for obtaining objective evidence.
This section also outlines the different types of audits, including internal and external audits conducted by regulatory bodies.
2. Navigating US and EU Audits
Medical device audits are mandatory for all device classes, but specific requirements vary depending on regulatory bodies and device classification.
In the EU and US, audits for medium to high-risk devices typically involve Notified Body audits for MDR/IVDR compliance and ISO 13485:2016 certification, FDA inspections for 21 CFR 820 compliance and manufacturing capability verification, and periodic surveillance audits.
Additionally, manufacturers are subject to unannounced and “for cause” inspections triggered by various issues.
3. Strategic Audit Preparation
Thorough preparation for an audit or inspection involves continuous auditing practices, mock audits, and self-identification of issues. Internal audits should be conducted rigorously, acting as rehearsals for external audits.
Mock audits, conducted by independent third parties, can reveal areas for improvement. Self-identifying issues and implementing corrective actions promptly demonstrates a proactive approach to compliance.
4. Selecting the Ideal Audit Host
When selecting an audit host, it’s crucial to choose someone who represents the company well, possesses in-depth knowledge of its operations and quality management system, and can handle pressure effectively.
The audit host is pivotal in ensuring a smooth and successful audit, so selecting the right individual is essential.
5. Document Readiness for Audits
To ensure a smooth audit process and avoid delays, organizations should pre-identify and readily have all necessary documents, including regulatory information, certificates, and records.
A centralized regulatory information management (RIM) system can significantly streamline the process by storing and linking to relevant documents from other systems.
6. Audit In-Action
During an audit, it is crucial to actively manage the process. The company host should introduce the organization and conduct a facility tour. While the auditor directs the audit, the host should assist and guide them throughout the process.
For unannounced inspections, a procedure should outline the reception and handling of such audits, including designating primary contacts and alternates.
Ideally, multiple company representatives should accompany the auditor, and they should not be left alone at any point. A “front room” team should transcribe every interaction and relay information to a “back room” team for support.
7. Best Information Sharing Practices
Employees should provide requested information to auditors but should consult with executives before sharing sensitive documents. Auditors should access information through photocopies or limited computer access.
Original documents can be presented but should not be kept by auditors. All information should be prepared, verified, recorded, and marked “Confidential” or “Proprietary” before being provided to auditors. An extra copy should be made for audit files.
8. Addressing Gaps in Information
Address missing or incorrect information by acknowledging the issue and discussing appropriate actions under the existing quality system. Be prepared to receive findings from any inspection and ensure that they are understood by both parties.
Address all findings diligently and respond to the regulatory body in charge of the audit with a satisfactory plan for correcting and preventing the recurrence of the identified issues.
Conclusion
In conclusion, medical device audits, though challenging, can be navigated successfully with thorough preparation and strategic responses.
Embrace the audit journey as an opportunity for continuous improvement, showcasing a commitment to compliance and the delivery of safe and effective medical devices.
Mastering medical device audits is not just a regulatory requirement – it’s a pathway to excellence in quality systems and product lifecycle management.
As technology advances across all healthcare fields, Software plays a significant role in all products. It is widely integrated into digital platforms serving both medical and non-medical purposes. Medical device software is one of three types of Software related to medical devices.
The other two types of medical device software include Software that is an integral part of the medical device (medical device software) and Software used in manufacturing or maintaining the medical device.
Software as a Medical Device Introduction
The International Medical Device Regulators Forum (IMDRF) defines SaMD as “software intended for one or more medical purposes that perform those purposes without being part of a hardware medical device.”
FDA defines SaMD as “Software that meets the definition of a device in 181 section 201(h) of the FD&C Act and is intended to be used for one or more medical purposes without being part of a hardware device.”
The use of SaMD is experiencing a steady rise, with its application extending to various technology platforms such as medical device platforms, commercial “off-the-shelf” platforms, and virtual networks, among others.
This kind of software was previously referred to as “standalone software,” “medical device software,” or “health software” by industry professionals, international regulators, and healthcare providers, often leading to confusion with other software categories.
How Do I Know if My Product is SaMD?
As a member of the International Medical Device Regulatory Forum (IMDRF), the FDA recognizes the structural similarity between the two organizations’ definitions of SaMD. The definitions provided by the FDA and IMDRF highlight two criteria that must be satisfied for software to be designated as SaMD.
To evaluate if the Software is a medical device, it is important to assess its compliance with the regulatory authority’s definition. The IMDRF emphasizes that the Software must be “intended for one or more medical purposes”. On the other hand, the FDA references FD&C, or the Federal Food, Drug, and Cosmetic Act, Section 201(h), which outlines the definition of a device. This section defines a device as follows:
According to Section 201(h) of the FD&C, a device encompasses various articles such as tools, implements, instruments, machines, devices, appliances, in vitro reagents and other similar or related items, including components and accessories.
An article must also be legally recognised in the National Formulary, the United States Pharmacopoeia, or an analogous revision in order to meet the requirements of Section 201(h) of the FD&C Act’s definition of a device.
The product is intended to be used in the diagnosis of disease or other conditions as well as in the treatment, mitigation, treatment, or prevention of disease in people or animals, according to the definition of a device under FD&C Section 201(h).
In addition, a product must be intended to change any structure or function of the human or animal body without compromising its primary function for it to qualify as a device under Section 201(h) of the FD&C Act.
In addition, the definition of a device in Section 201(h) of the FD&C Act states that it must not achieve its primary purpose by chemical action in or on the human or animal body, nor must it rely on metabolism to achieve its purpose.
Note that the term “device” does not include software features that are excluded by Section 520(o). To apply this definition, it is essential to establish the intended use and indications for the use of your product. To refresh your understanding,
The intended use of a device refers to its designated purpose or the specific function for which the device is intended to be utilised. In other words, it defines the intended or intended application of the device
Indications for use pertain to the diseases or conditions that a device is designed to diagnose, treat, prevent, cure, or mitigate. These indications specify the target patient population and provide insights into why the device would be used on individuals with those diseases or conditions
Once the intended use and indications for the use of your product are defined, the second and third points in the FDA’s definition of a medical device will help determine whether your product falls under the regulatory scope of a medical device. These criteria will clarify whether your product meets the requirements for medical device regulation.
If you intend to market your software product in the United States, I recommend carefully reviewing the FDA’s guidance on Policy for Device Software Functions and Mobile Medical Applications.
This guidance provides valuable insights into the specific software functions that the FDA classifies as medical devices and functions that are not considered medical devices and, therefore, not subject to FDA regulation. Familiarizing yourself with this guidance will provide you having a thorough awareness of the regulatory environment for software products in the medical field.
If you’re still unsure about whether your product qualifies as a medical device, contacting the FDA directly for clarification is safest. Contacting the FDA now will provide reliable advice and ensure you receive accurate information tailored to your specific product and situation.
Is My Software Considered SaMD or SiMD?
When a product is found to fit the definition of a medical device, the second portion of the IMDRF and FDA definition of software as a medical device (SaMD) must be taken into account.
According to the IMDRF definition, Software must be used for its intended purpose only and must not be an integral part of a physical medical device.
According to the IMDRF, the FDA makes it clear that Software as a Medical Device (SaMD) is not a part of a hardware device and is instead intended for standalone usage for one or more medical purposes.
This further limit the scope of Software as a medical device (SaMD), as the Software used to operate or control a hardware device does not fulfil the requirements to be categorised as a SaMD. Instead, this type of Software is referred to as SiMD or “software in a medical device.”
Here are some examples of Software that assist in operating a hardware medical device, which falls under the category of Software in a Medical Device (SiMD) and not Software as a Medical Device (SaMD):
Software that controls the inflation or deflation of a blood pressure cuff
Software that controls the delivery of insulin on an insulin pump
Software used in the closed-loop control of a pacemaker.
“firmware,” or “micro-code,” indicating their classification as SiMD rather than SaMD.
To qualify as SaMD, a product must have standalone Software that independently carries out the functions defining it as a medical device, distinct from any associated hardware. The IMDRF guidance on “Possible Framework for Risk Categorization and Corresponding Considerations” further provides additional insights and clarifications regarding the SaMD definition.
1. SaMD is a medical device, including in-vitro diagnostic (IVD) medical devices.
2. SaMD can run on general-purpose computing platforms not specifically designed for medical purposes.
3. “Without being part of” means the Software is unnecessary for a hardware medical device to achieve its intended medical purpose.
4. Software intended to drive a hardware medical device does not meet the definition of SaMD.
5. SaMD can be combined, such as being integrated as a module, with other products, including medical devices.
6. SaMD can interface with other medical devices, including hardware medical devices, other SaMD software, and general-purpose Software.
7. Mobile apps that meet the defined criteria are considered SaMD.
Although it is crucial to distinguish between Software as a Medical Device (SaMD) and Software in a Medical Device (SiMD), both types of Software adhere to many common standards for development, including the global standard for software lifecycle procedures is IEC 62304. lifecycle processes.
If your Software falls under the category of SiMD, you will still find the guidance documents and standards outlined in this guide valuable and applicable.
What are Some Examples of SaMD?
1. Software that enables a mobile device to view diagnostic images from MRI, ultrasound, or X-ray scans.
2. Software that utilizes image processing techniques to aid in detecting breast cancer.
3. Software that diagnoses a medical condition by analyzing data from the tri-axial accelerometer on a smartphone.
4. Software that collects real-time patient data monitored by a healthcare professional and utilized to develop treatment plans.
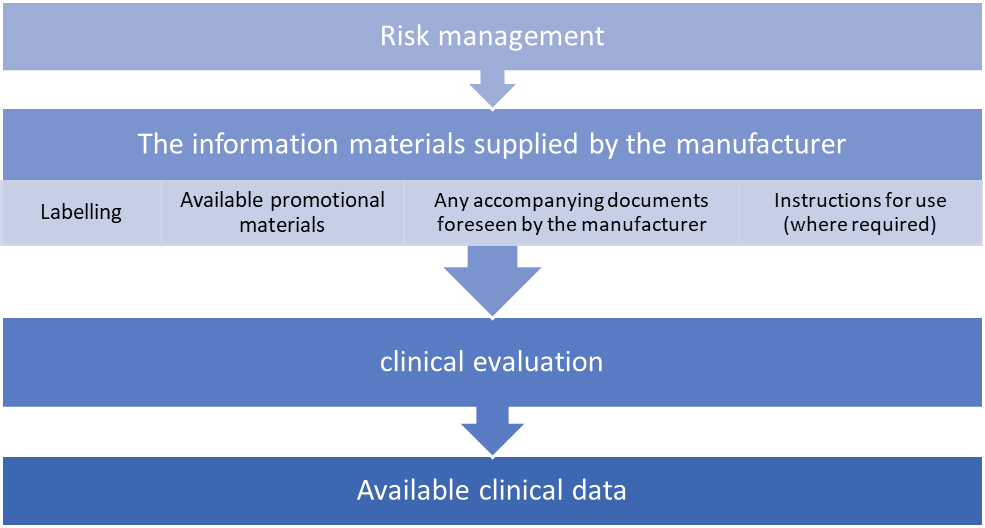
Clinical Evaluation
Clinical evaluation is a methodical and well-organized process that creates clinical evidence by continuously creating, collecting, analyzing and evaluating clinical data on SaMD to create clinical trials that review the clinical context and performance indicators of SaMD.
The quality and scope of the clinical assessment is based on the SaMD function for the clinical objective, which also ensures that the SaMD score is clinically valid and can be used consistently and predictably.
3 Clinical Evaluation Software
To qualify the software, the following three criteria must be met. To qualify your software, you must meet the three criteria outlined below.
Valid Clinical Association of a SaMD
Analytical/Technical Validation of a SaMD
Clinical Validation
1. Valid Clinical Association of a SaMD
Verifying that your software’s output corresponds to the targeted clinical conditions is the main goal here. Use of secondary data analysis, clinical trials (new data generated), professional society guidelines, original clinical research, literature searches, and/or secondary data analysis are all options for carrying out that task. All SaMD should show a reliable clinical association.
2. Analytical/Technical Validation
Does your software correctly process input data to generate accurate, dependable, and precise output data?” is the question we are attempting to answer in this context. Develop supporting documentation that demonstrates your SaMD’s output met your expectations in terms of technicality.
This action is being assessed by a manufacturer as part of the software’s validation and verification phase (V&V).
3. Clinical Validation
SaMD has been evaluated in your target population and for your intended use; users are able to achieve clinically significant results through consistent and dependable use.
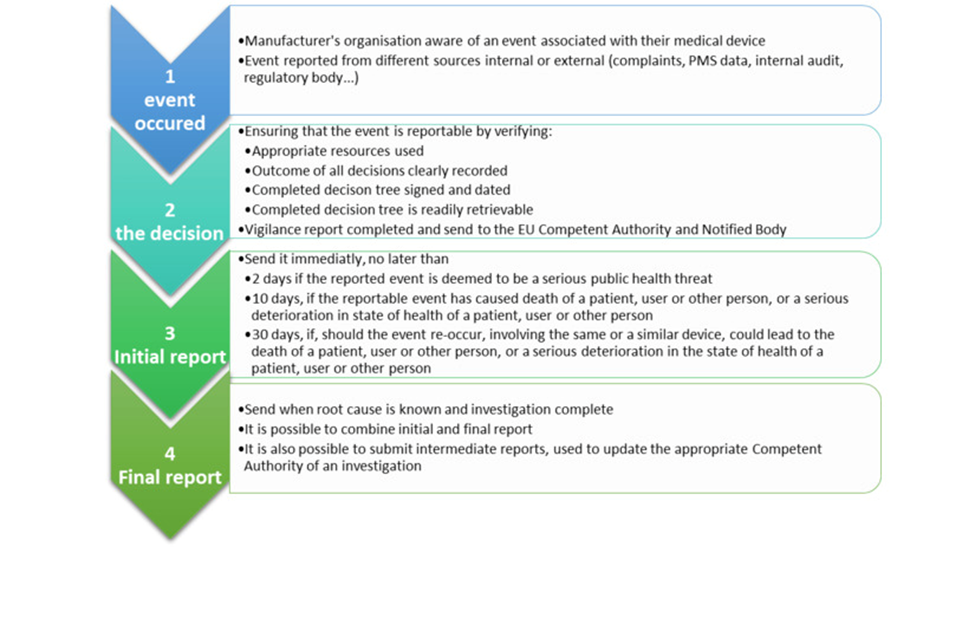
According to European Union Medical Device Regulation (EU MDR) the term “Vigilance” is the identification, reporting and trending of serious incidents and the conduct of safety-related corrective actions (Market surveillance and vigilance).
An ‘incident’ as per article 2(64) MDR is any malfunction or deterioration in the characteristics or performance of a device made available on the market, including use-error due to ergonomic features, as well as any inadequacy in the information supplied by the manufacturer and any undesirable side-effect.
A ‘serious incident’ as per Article 2(65) MDR is an incident as outlined in Article 2(64) MDR that has in addition, either led to or has the potential to lead to significant health or public health outcomes.
In essence, serious incidents refer to a particular category of incidents that have caused, have the potential to cause, or may cause death or severe harm to a patient, user, or any other person.
These incidents can also pose a significant threat to public health. As per Article 87(1) to (5) of the MDR, it is the responsibility of the manufacturer to report such serious incidents to the appropriate regulatory authority.
Any incident which meets all three basic reporting criteriaA to C listed below is considered a serious incident and must be reported to the relevant competent authority:
An incident has occurred
Examples:
A malfunction or deterioration in the characteristics or performance of the device, e.g. a device that fails or is losing its ability to achieve its intended purpose when used as indicated in the information supplied by the manufacturer
A deterioration in the characteristics of the device that is related to manufacturing errors, e.g. sterilization process failures
A use error due to ergonomic features, e.g. a use error caused by a mismatch between the user interface and the physical or medical condition of the intended user
Any inadequacy in the information supplied by the manufacturer, e.g. insufficient information on how to maintain, adjust or calibrate the device in the instructions for use
Unclear instructions in the labelling or the manufacturer’s instructions for use
Undesirable side-effects e.g. allergic skin reactions such as allergy to nickel or wound therapies
Incident directly or indirectly led, might have led or might lead to any of the outcomes of a serious incident such as death of a patient, user or other person; temporary or permanent serious deterioration of a patient’s, user’s or other person’s state of health; or a serious public health threat. Read more on the concept of vigilance in EU MDR on our website.
Examples of serious public health threats:
Contagious illnesses, such as HIV, Ebola, Zika virus, SARS and COVID-19
Events involving high risk of exposure to a disease (e.g., cancer) after use of a medical device, which affects a specific patient population (diabetics, cardiac patients, etc.) or a vulnerable population (children, pregnant women, etc.)
Exposure to toxic compounds with a potentially negative/harmful effect on humans
Widespread distribution of falsified or incorrectly labelled devices leading to multiple serious incidents, e.g. distribution of non-sterile devices labelled as sterile
Cyberattack related to life supporting or life-saving devices
Causal relationship between the serious incident and the manufacturer’s device has been established or is reasonably possible or suspected.
User, Use error and Abnormal use of a medical device
According to Article 2(37) of the MDR, a “User” refers to any healthcare institution, healthcare professional, or lay person (such as a caregiver or patient) who uses a device, as well as those who install or maintain the device. The term “operator” may also be used to refer to the user of a device.
A “use error” occurs when the actions or inactions of the user while using the device result in a different outcome than intended by the manufacturer or expected by the user.
This can be due to various reasons such as lack of attention, memory lapses, mistakes during device use, or insufficient knowledge or understanding of device use.
Such use errors are not considered incidents, but if they are caused by ergonomic features of a device, they are considered incidents and must be reported under Article 87(1) of the MDR in case of serious incidents.
“Abnormal use” refers to the intentional violation of the intended use of a device. This may involve deliberate acts or omissions that deviate from the normal use of the device and cannot be controlled by the manufacturer.
For example, when a doctor uses a device for an indication that is not specified in the manufacturer’s instructions for use.
“Use-error due to ergonomic features” refers to use errors caused by the physical features of a device that are designed to ensure safe, effective, and efficient interaction between the user and the device.
These errors may occur when there is a mismatch between the device features and the user profile or the environment in which the device is used.
Ergonomic features can be described as the physical features of a device that are designed to facilitate and ensure that the interaction between the user and the device is safe, effective and efficient.
Ergonomic features include components such as measurement and monitoring features, display scales, alarms, software menus, and other factors related to the user interface.
Reporting under MDR
The MDR mandates that the timeline for reporting serious incidents should be based on the severity of the incident. These reporting periods are counted as calendar days.
Typically, the reporting period begins the day after the manufacturer becomes aware of a potentially serious incident. The awareness date, which is considered day 0, is when the manufacturer first receives information about the incident, not after any investigation has been conducted.
The manufacturer’s awareness date is determined as the day when any employee or representative of the manufacturer’s organization first receives information, such as a complaint, about the potentially serious incident.
This can be illustrated with an example:
On June 1, 2022, a manufacturer receives a complaint but decides that it does not meet the criteria for a serious incident, so they do not submit a Manufacturer Incident Report (MIR) to the relevant authority.
However, on July 1, 2022, the manufacturer receives more information and reviews it, and now determines that the complaint is indeed a serious incident. The manufacturer must thus submit a MIR no later than July 16, 2022.
In the MIR, the manufacturer should provide the relevant dates in the following two fields:
‘Manufacturer awareness date’ – in this field, the initial awareness date of the incident should be inserted by the manufacturer.
‘General comments’ – in this field, the manufacturer should insert the date in which it received the information that determined that the incident is reportable
A ‘field safety corrective action (FSCA)’ is a corrective action taken by a manufacturer for technical or medical reasons to either prevent or reduce the risk of a serious incident, which is associated with a device that is made available on the market.
A Field Safety Corrective Action (FSCA) can involve various actions such as returning the device to the supplier or recalling it, exchanging the device, modifying it, retrofitting it with the manufacturer’s modification or design change, destroying it, giving advice on the device’s use, recommending inspections/examinations of the device by the user, making changes to the device’s software/firmware, and updating the device’s version (for example, rolling it back to an earlier version).
Example of an FSCA conducted by a manufacturer:
As a part of monitoring their products after they have been released to the market, the manufacturer has found a consistent issue with one of their devices.
If this issue could result in a serious problem and the affected devices are still being sold, the manufacturer is required to initiate a FSCA (Field Safety Corrective Action) to prevent or minimize the risk of such incidents.
This FSCA may involve making permanent or temporary changes to the device’s labeling or instructions for use, or recalling all of the affected devices from the market. The manufacturer must promptly inform the relevant authorities in the Member States where the FSCA is being carried out.
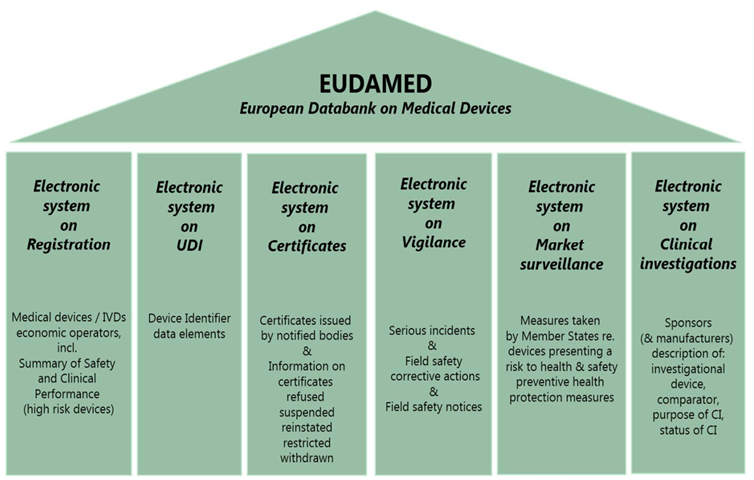
Vigilance Reporting in Eudamed
Eudamed is a recently established database by the European Union for medical devices, designed to gather all necessary information about devices that have been made available in the Union market.
The details of the database can be found in Article 33 MDR, while the rules regarding vigilance are described in Article 92 MDR. The Medical Device Coordination Group (MDCG) guidance provides suggestions on temporary measures that can be used to comply with certain MDR requirements related to Eudamed and information sharing.
These alternatives enable Member States and other stakeholders to fulfill their obligations under the MDR until the complete operation of the database.
Periodic Summary Report
A “Periodic summary report” (PSR) is another way for manufacturers to report serious incidents involving the same device or device type in a consolidated manner.
This reporting method is used when the manufacturer agrees with the relevant national competent authority that coordinates periodic summary reporting.
The PSR is based on certain criteria, which includes situations where the root cause of the incidents has been determined, a Field Safety Corrective Action (FSCA) has been implemented, or where the serious incidents are commonly known and documented.
A ‘common and well documented serious incident’ as referenced in Article 87(9) MDR, must be clearly identified in the manufacturer’s risk analysis and should have led to incident reports, which have been assessed by the manufacturer and the relevant competent authority.
FAQs
Who is responsible for vigilance activities related to medical devices?
The manufacturer of a medical device is responsible for vigilance activities related to that device. This includes monitoring the device’s performance, investigating any incidents or complaints related to the device, and reporting any adverse events to the relevant authorities.
What is a trend report?
A trend report is a summary of adverse event data related to a particular medical device or group of devices. It is used to identify patterns or trends in adverse events and to assess the potential risks associated with the device.
What are the basic reporting criteria for a serious incident?
· An incident (Article 2(64) MDR) has occurred · The incident directly or indirectly led, might have led or might lead to any of the outcomes of a serious incident (Article 2(65) MDR) · A causal relationship between the serious incident and the manufacturer’s device has been established, is reasonably possible or suspected
A general device description, including any information on any planned variants
Design drawings, details on the planned method of manufacture, diagram of components, sub-assemblies, circuits etc
Descriptions and explanations are required to understand the abovementioned drawings and diagrams and the operations of the product
Results of risk analysis and a list of standards that are applied in full or part (Standards are referred to in Article 5 MDD)
Description of the solutions adopted to meet the essential requirements of the Directive if standards have not been applied fully. (Standards are referred to in Article 5 MDD)
Sterility information, description, and methods of use of sterile products
Results of design calculations and inspections carried out
If the device is to be connected to other device(s) to operate as intended, then there must be proof provided to indicate that it conforms to the essential requirements when connected to any such device(s) having characteristics specified by the manufacturer
Test Reports
Clinical Reports wherever applicable and Clinical data as per Annex X of MDD
Label and Instructions for use
MDR
All Technical documentation requirements of MDD must be presented for the MDR alongside the below additional list:
Device Description and Specifications
Basic UDI-DI
the intended patient population and medical conditions to be diagnosed
contra-indications and warnings
principles of operation of the device and its mode of action
the rationale for the qualification of the product as a device
the risk class of the device and the justification for the classification rule(s) applied
an explanation of any novel features
A description of the accessories for a device, other devices and other products that are not devices intended to be used in combination with it.
a description or complete list of the various configurations/variants of the device
a general description of the key functional elements, e.g., its parts/components
a description of the raw materials incorporated into key functional elements and those making either direct contact with the human body or indirect contact with the body
Technical specifications
Reference to previous and similar generations of the device
Information to be supplied by the manufacturer
A complete set of labels or labels on the device and on its packaging
The instructions for use in the languages accepted in the country of sale
Design and Manufacturing Information
information to allow the design stages applied to the device to be understood
complete information and specifications, including the manufacturing processes and their validation, their adjuvants, the continuous monitoring and the final product testing
identification of all sites, including suppliers, sub-contractors and manufacturing sites.
General Safety and Performance Requirements
The general safety and performance requirements that apply to the device and an explanation as to why others do not apply
The method or methods used to demonstrate conformity with each applicable general safety and performance requirement
the harmonised standards, CS or other solutions applied
the precise identity of the controlled documents offering evidence of conformity with each harmonised standard, CS or other method applied to demonstrate conformity
Benefit-Risk Analysis and Risk Management
The benefit-risk analysis, the solutions adopted, and the results of the risk management
Product Verification and Validation
The documentation shall contain the results and critical analyses of all verifications and validation tests and/or studies undertaken to demonstrate the conformity of the device with the requirements of this Regulation.
Pre-Clinical and Clinical data
Results of tests
If applicable: biocompatibility report, physical, chemical and microbiological characterisation, electrical safety and electromagnetic compatibility, software verification and validation,
stability, including shelf life,
performance and safety
Where applicable, conformity with the provisions of Directive 2004/10/EC of the European Parliament and of the Council (1) shall be demonstrated
Where no new testing has been undertaken, the documentation shall incorporate a rationale for that decision
the clinical evaluation report and its updates and the clinical evaluation plan
the PMCF plan and PMCF evaluation report, and if not applicable, justification of why a PMCF is not applicable
Additional information required in specific cases
Data of the tests conducted to assess safety, quality and usefulness on:
Medicinal product used
Medicinal products derived from human blood or human plasma
Tissues or cells of human or animal origin or their derivatives
Substances or combinations of substances that are intended to be introduced into the human body and that are absorbed by or locally dispersed in the human body
CMR (carcinogenic, mutagenic, or toxic for reproduction) substances
Endocrine-disrupting substances
A description of:
Sterility or defined microbiological condition to be maintained
Methods used in devices with measuring functions to ensure the accuracy as given in the specifications.
Combination/configuration of devices connected to other devices (s) to operate as intended, including proof that it conforms to the general safety and performance requirements when connected to any such device(s) having regard to the characteristics specified by the manufacturer
Post Market Surveillance
Post-market surveillance plan drawn up in accordance with Article 84
Post Market surveillance plan shall address:
Information concerning serious incidents, including information from PSURs, and field safety corrective actions
Records referring to non-serious incidents and data on any undesirable side-effects
Information from trend reporting
Relevant specialist or technical literature, databases and/or registers
Information, including feedback and complaints, provided by users, distributors and importers
Publicly available information about similar medical devices
The post-market surveillance plan shall cover at least:
A proactive and systematic process to collect any information
Effective and appropriate methods and processes to assess the collected data
Suitable indicators and threshold values shall be used in the continuous reassessment of the benefit-risk analysis and the risk management
Effective and appropriate methods and tools to investigate complaints and analyse market-related experience collected in the field
Methods and protocols to manage the events subject to the trend report
Methods and protocols to communicate effectively with competent authorities, notified bodies, economic operators, and users
Reference to procedures to fulfil the manufacturer’s obligations
Systematic procedures to identify and initiate appropriate measures, including corrective actions
Effective tools to trace and identify devices for which corrective actions might be necessary
A PMCF plan, or a justification as to why a PMCF is not applicable
The PSUR referred to in Article 86 and the post-market surveillance report referred to in Article 85
Disclaimer: Regulations/legislations are subjected to changes from time to time and the author claims no responsibility for the accuracy of information.
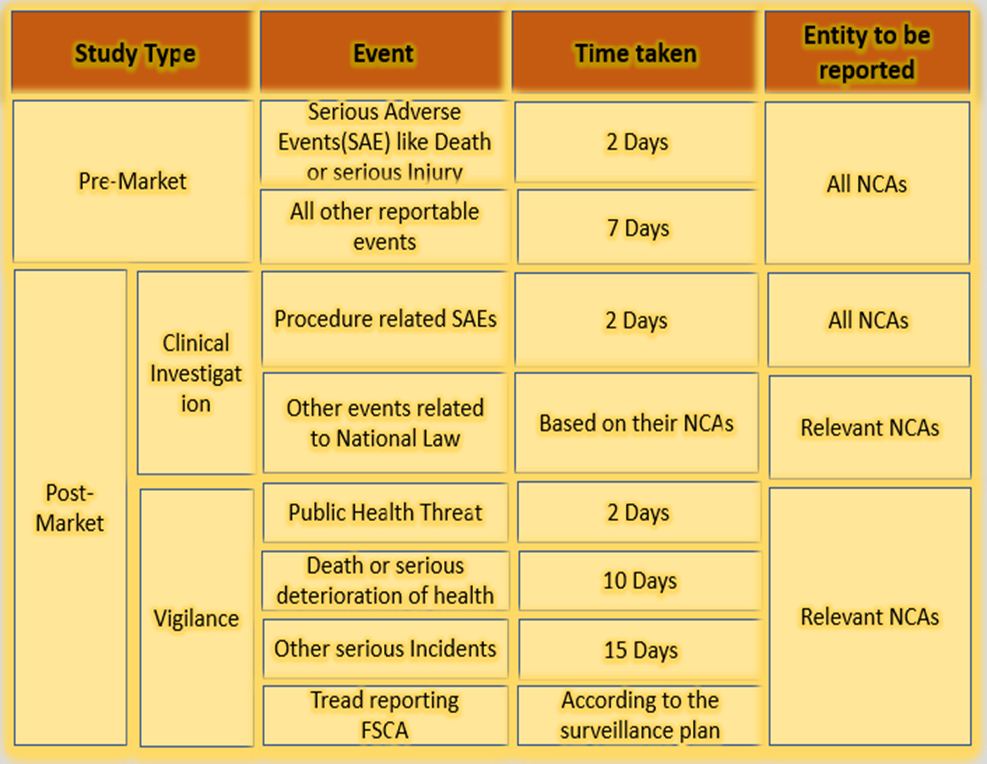
Under EU MDR 2017/245 Article 73, the sponsor shall report to all member states about the clinical investigations undertaken by the medical devices through an electronic system in case of any serious adverse events, device deficiency which leads to adverse events or any novel findings with regards to the event.
The submission period may vary depending on the severity of the events. An initial incomplete report followed by the complete report shall be provided.
The reporting procedures are applicable for both Pre-market clinical investigations and Post-market clinical follow-up. Pre-market Clinical Investigation is conducted for non-CE marked medical devices, CE Marked devices outside the intended use as per Articles 62 and 74(2).
From 26th May 2021, a new template shall be used for reporting clinical investigation, which must be filled or updated with all adverse events, or any novel findings or occurrences reported.
This report must be communicated to all National competent authorities. As per Article 34(3), the transition of reports will be made available on EUDAMED when it is completely available and fully functional. The overview of the table format to be used by the sponsor is given below.
Types of clinical investigation:
As per Article 80(2,3), the clinical investigation may be used for the investigational device for the conformity procedure.
As per Article 80(5,6), the PMCF investigation in case of serious events or invasive procedures
Other clinical investigations regarding National Legislation for Safety reporting
In addition to the above-mentioned clinical investigations, there are some exceptions other than the serious adverse events in the causal relationship between the event and the preceding investigational procedure like
Other relationships unrelated to the preceding investigational procedures need not be reported.
Few events in the relationship between the procedure or the device, as stated in IFU, shall be taken in context with the vigilance as per Article 87-90
A procedure mandated by the Clinical Investigation Plan that occurred prior to (or at the same time as) the major adverse event is referred to as a “preceding investigational procedure.”
Both the investigational device application process and any additional onerous or intrusive procedure(s) determine whether the study is subject to notification requirements under MDR article 74.
Reporting Events to other Member States/ Third party countries
The sponsors shall report all the clinical investigations to those Member states. In the case of many Clinical investigations conducted for the same device, the specific CIP code (Clinical Investigation Plan Code) is to be reported to the specific Member state.
Reports from Third party countries shall be received through the National Competent Authorities as soon as the clinical investigation is authorized, and all other events occurring in third-party countries need to be reported.
Reported by whom? To Whom and When?
The sponsor of the clinical trial, the manufacturer, the legal agent, or another person or entity, must disclose any reportable events to the National Competent Authorities (NCA).
On the Commission’s website, a list of the phone number of these operational NCAs is published. Member states may also require a supplemental report to the ethics committee(s).
Regarding reporting timelines, investigators must inform sponsors as soon as possible and no later than three days after the investigation site study employees become aware of the incident.
The sponsor must adhere to the following reporting timelines based on the type of clinical study and the severity of the adverse event.
Timeline for reporting events
The Investigator’s Brochure, the Clinical Investigation Plan, or the Risk Analysis Report should be consulted during the causality assessment process.
All foreseeably serious adverse events and potential risks need to be listed and evaluated during the clinical investigation. MDCG 2020-10/1 provides clear guidance on harmonizing reports, where the serious adverse events are further classified into the following four levels of causality (investigational device, comparator, procedure):
Not related: No relationship to the events related to the investigational device, an event that does not affect the device procedure, the serious adverse event that does not follow any known pattern but is related to other causes like concurrent illness, clinical condition or other risk factors
Possible: The relationship between investigational procedures, devices, or comparator is tenuous but cannot be excluded. Alternative explanations, such as an underlying or concurrent sickness, clinical condition, or course of therapy, are also conceivable. Cases where the relationship cannot be determined or where no information is known should also be categorized as possible.
Probable: The association with the application of the investigational device, comparator, or procedures appears pertinent, and/or another cause cannot adequately explain the incident.
Causal relationship: The serious adverse event associated with an investigational device, comparator, or procedure when the event has known side effects which involve a body site or organ or if the event follows a responsive pattern or other possible causes that can cause harm to the subject.
The investigator must identify the relationship between the serious adverse event and the procedure, as they can be related to each other, or they can be related only to the procedure or device itself.
The health care provider can carry out the above procedure as they manage or handle the medical devices.
The reporting form is study-specific and only applies to a particular clinical investigation as identified by a specific clinical investigation plan. The reporting form should be completed in English.
The report file must be compatible with Microsoft Excel before being delivered to the NCAs.
What is a clinical investigational plan for medical devices?
A clinical investigational plan is a document that states the objectives, design, procedure, monitoring and method of recording the clinical investigation for the medical devices. The ability to attain the intended purpose of the medical devices, as claimed by the manufacturer, is called clinical performance.
What is a clinical safety report?
All the data related to the safety information on adverse events, serious adverse events, and other risk factors of the medical devices in the user facility must be recorded into a form which is termed a clinical safety form/report. This report gives awareness to everyone in the workplace.
Why is safety reporting important in clinical trials?
To ensure the safety and quality of the clinical trials conducted by the investigator. These safety reports must be submitted to the respective NCAs through the sponsors during the entire lifecycle of the product trial.
A medical device’s intended use and inherent risks must be considered when determining its MDR classification. Class I devices pose less risk to patients and end users, as under the previous MDD. The new Regulation EU MDR 2017/745 has added extended rules, leading some devices to fall under Class IIa, IIb, or even III.
This document is intended to guide Class I medical device manufacturers (excluding custom-made devices) that sell products bearing their name or trademark on the Union market to comply with MDR requirements. This guidance should also apply when an importer, distributor, or any other legal entity takes on the obligations owed by manufacturers.
The class I manufacturer must also hire a Notified Body (NB) to perform a conformity evaluation if medical devices are to be provided sterile, have measuring functions, or are reusable surgical instruments. For the NB to be qualified to conduct such a conformity assessment, the specific medical device in question must come within the purview of the Notified Body’s designation.
Large and mid-sized medical device manufacturers must also properly designate at least one individual in charge of regulatory compliance, but micro and small companies can function better by having a regulatory expert.
It is also important to designate an authorised representative residing within the EU if the manufacturer is registered outside of the EU. A foreign manufacturer must specifically issue a mandate outlining the representative’s duties and authority. It is also crucial to note that not all responsibilities associated with the medical equipment can be assigned.
The manufacturer must adequately preserve all the records required to verify compliance with the relevant requirements so they may be made available to the regulatory body upon request.
Steps to placing Class I medical devices on the market:
Manufacturers must ensure compliance with each of the following requirements if they plan to commercialise Class I medical devices. Please be aware that some of the outlined requirements are interconnected and can be completed in a different sequence than that which is shown.
The manufacturer will carry out a gap analysis for Class I devices that have already been released onto the market in compliance with the MDD to ensure that all of the below-listed requirements were satisfied at the time the MDR was applied.
The whole procedure includes a set of mandatory steps, including:
Step 0: Integrate MDR in the Quality Management System (QMS)
The manufacturer’s QMS should be appropriately linked with the standards outlined in the MDR. To ensure compliance with the following requirements, this will enable the correct assessment or decision to be taken and the appropriate documented evidence to be produced.
Step 1: Confirm product as a medical device
The intended purpose and the product’s mode of action are reviewed as per Article 2(1) of the MDR to ensure that the product qualifies as a medical device. If a product is assigned multiple intended purposes, it will qualify as a medical device only if all the intended purposes are covered in Article 2(1).
In the case of products identified as more than one category, the relevant legislation requirements will have to be followed. Despite not being medical devices in and of themselves, accessories to medical devices are subject to MDR regulations and qualify as devices under the MDR definition.
However, the MDR does not include accessories to devices covered by it as per Annex XVI of the MDR.
Step 2: Confirm product as a Class I medical device
The product has to be confirmed if it can be classified as a class I device as per Annex VIII of the MDR. Products previously classified as class I under MDD should be reviewed according to the classification rules of MDR to confirm if reclassification is required.
The MDD guideline cannot be used for devices that were moved from Class I to higher risk classes by the adoption of the MDR. The intended use of the device and any inherent risks related to the period of use, the part of the body, whether it is active or not, and whether it is invasive or non-invasive will determine how the classification standards are applied.
The classification rule with the highest class should be applied if the device in question falls under the purview of more than one classification rule due to its attributes.
Step 3: Procedures before placing on the market
a) Meet the General Safety and Performance Requirements (GSPR)
The devices will adhere to the general safety and performance standards outlined in Annex I of the MDR, considering the intended purposes that their manufacturers had provided.
The Risk management system is a continuous iterative process throughout the entire lifecycle of the product, established by the manufacturer that will enable the identification and analysis of the risks related to each device, the estimation and assessment of the risks associated with those risks, the elimination or control of residual risks, and the evaluation of the adopted measures based on the data gathered from the PMS system.
When a harmonised standard is in place, but a manufacturer chooses to use another reference, implementing that reference should, at the very least, ensure the same level of performance and safety.
Compliance with the relevant harmonised standards will give rise to a presumption that the MDR’s requirements, or portions of them, are also complied with.
If there are available standard specifications, the manufacturer must adhere to them unless they can adequately demonstrate that they have chosen a solution at least as effective and safe. The clinical evaluation processes, risk management, and PMS must be interdependent and updated regularly.
b) Conduct clinical evaluation
As part of the MDR’s technical documentation requirements, all devices—regardless of risk classification—require a clinical evaluation. The level of clinical evidence required to show compliance with the pertinent general safety and performance requirements listed in Annex I, which is obtained by considering the characteristics of the device and its intended purpose, shall be specified, and justified by the manufacturer.
Manufacturers must prepare, carry out, and record a clinical evaluation in compliance with Article 61 and Part A of Annex XIV to accomplish this.
Conformity to Annex I requirements can only be assumed when the following items are aligned with each other.
Consideration of available alternative treatment options, the incorporation of clinical data and the acceptability of the benefit-risk ratio are required for carrying out a clinical evaluation.
Additional clinical data will be acquired or developed by clinical investigations if the currently available clinical data are insufficient to establish compliance with the MDR.
If the clinical data currently available for a device that is currently certified under Directive 93/42/EC is insufficient to show compliance with MDR, then post-market clinical follow-up studies of the device may be used to gather more clinical data.
Even data from the general post-market follow-up may occasionally be enough to close the difference.
a) Prepare technical documentation
The manufacturer is responsible for creating and maintaining the technical documentation to prove that their products adhere to the MDR’s technical specifications. Under Annex II and III, this technical documentation must be prepared before the EU declaration of compliance is drawn.
The manufacturer will develop and provide the technical documentation and, if applicable, it’s summary in a way that is unambiguous, clear, well-organised, and easy to search.
The manufacturer shall provide the CA, the authorised representative (where applicable), and NB with access to the technical documents (when applicable).
After reviewing the general safety and performance standards, as well as the pertinent technical provisions of the MDR, the technical documentation will be developed.
b) Request Notified Body involvement
Class I devices do not require the engagement of an NB for MDR compliance. But the manufacturer must follow the guidelines outlined in Chapters I and III of Annex IX or Part A of Annex XI of MDR when the product is a sterile equipment, measuring instrument, or reusable surgical tools.
For the relevant codes and corresponding types of devices as established by Regulation (EU) 2017/2185, manufacturers may select any NB designated in accordance with the MDR.
c) Prepare Instructions for Use and Labelling
Any safety and performance information required for a device’s safe usage and identification of the device should be provided along with the device by the manufacturer and/or the authorised representative, considering the possible users’ training and knowledge.
This data is included on the label, in the device packing, and in the instructions for use. Class I devices do not need instructions for use if they may be operated effectively and securely without them, deviating from the general rule.
Since Class Ir devices will need instructions for reprocessing (cleaning and sterilisation), an exception is most likely proposed.
Labelling and instructions for use must be written in accordance with national language regulations. There will be versions of the labelling and IFU in the technical documentation (in each pertinent national language).
The requirements regarding the information to be supplied with the device will be found in Annex I, Chapter III (23).
Step 4: Check compliance with general obligations for manufacturers
The manufacturer shall ensure that the general obligations for manufacturers outlined in Article 10 are met before releasing a device for sale.
Implementing a suitable QMS that will most effectively assure compliance with the MDR, such as through an internal audit, will receive special consideration. The QMS shall be documented, applied, maintained, updated regularly, and developed continuously, and it will at least include the following elements:
A strategy for regulatory compliance
Identification of applicable general safety and performance requirements and exploration of options to address those requirements
Responsibility of the management
Resource management, including selection and control of suppliers and sub-contractors
Risk management
Clinical evaluation, including post-market clinical follow-up (PMCF)
Product realisation, including planning, design, development, production and service provision
Verification of the UDI assignments
Setting up, implementation and maintenance of a post-market surveillance system
Handling communication with competent authorities, notified bodies, other economic operators, customers and/or other stakeholders
Processes for reporting serious incidents and field safety corrective actions in the context of vigilance
Management of corrective and preventive actions and verification of their effectiveness
Processes for monitoring and measurement of output, data analysis and product improvement.
According to applicable Union and national law, either natural or legal persons may seek compensation for harm brought on by a defective equipment.
In order to protect themselves against potential responsibility under Directive 85/374/EEC, manufacturers must take precautions that are commensurate to the risk class, type of device, and size of the business. These steps must not compromise further protective measures under national legislation.
Step 5: Draw-up the EU Declaration of Conformity
The process by which the manufacturer, who complies with the standards established by Article 52(7), certifies that the devices in question comply with the requirements of the MDR that apply to them is referred to in Article 19 as the EU declaration of conformity.
The CA will have access to the declaration of compliance, which must include, at a minimum, all of the data referred to in Annex IV. The manufacturer will regularly update the EU declaration of conformity and translate it into the official language(s) required by the Member States where the product is sold.
Suppose a device is subject to additional Union laws in addition to the MDR that also call for an EU declaration of conformity. In that case, the manufacturers will create a single EU declaration of conformity that refers to all applicable Union laws.
By creating the EU declaration of conformity, the manufacturer accepts liability for the device’s regulatory compliance with all relevant Union legislation.
For class Ir, Im, and Is devices, the manufacturer must obtain an EC certificate from NB per Annex IX, Chapters I and III, or Annex XI, Part A before applying a CE mark.
Step 6: Affix the CE marking
All Class I medical devices on the market must display the CE conformity label, which must be visible, readable, and permanent. It may be applied to the item or its sterile packaging. The CE marking must be applied to the package in cases where such affixing is impossible or unwarranted due to the nature of the device. The CE certification must be visible on all sales packaging and the instructions for use.
Placing the CE mark on a medical device signifies that it complies with all applicable safety and performance standards and is approved for marketing in the EU. The CE marking will be accompanied by the identification number of the relevant NB in the case of Class I medical devices placed on the market in a sterile condition, devices with measurement functions, and/or reusable surgical tools.
Affixing marks that could lead third parties astray about the significance of the CE marking is banned. Other extra markings may be applied to the product, the packaging, or the usage instructions, but they must not obscure or obstruct the CE marking.
The CE marking format will follow Annex V requirements. The minimum dimensions required for the CE mark may not apply to very small devices.
Step 7: Registration of devices and manufacturers in Eudamed
A streamlined approach might be used for medical devices that were previously put on the market per the Directives, provided that the manufacturer evaluates the gap and ensures that all regulations are properly followed.
A Class I medical device manufacturer must register the product with Eudamed before putting it on the market. Suppose the information listed in Section 1 of Part A of Annex VI has not already been registered in accordance with Article 31. In that case, the manufacturer must submit it to the electronic system in order to register the device. The information referred to in Section 1 of Part A of Annex VI will be given to that electronic system before applying to the NB in situations where the conformity assessment method necessitates the engagement of an NB in accordance with Article 52.
Following the CA’s validation of the manufacturer’s data in Eudamed, the manufacturer will receive an SRN from the aforementioned electronic system. To fulfil its duties under Article 29, the manufacturer will use the SRN when submitting an application to an NB for a conformity assessment and to gain access to Eudamed.
The registration of a device in Eudamed by the manufacturer includes:
Assigning a UDI-DI (with a Basic UDI-DI) as described in Part C of Annex VI to the device in accordance with the issuing entity’s policies mentioned in Article 27(2) and adding the UDI-DI (with a Basic UDI-DI) to the UDI database along with the other core data elements pertaining to that device mentioned in Part B of Annex VI.
Entering the data referred to in Section 2 of Part A of Annex VI, excluding Section 2.2 thereof, or, if previously provided, validating the data, and then keeping the data current up to date.
This document summarises the information presented above and outlines how Class I medical devices should be introduced to the EU market. The guidance’s scope also includes reusable surgical instruments (Class Ir), sterile medical devices (Class Is), and devices with measurement capabilities. These medical devices all need a Notified Body to be involved in pre-marketing procedures.